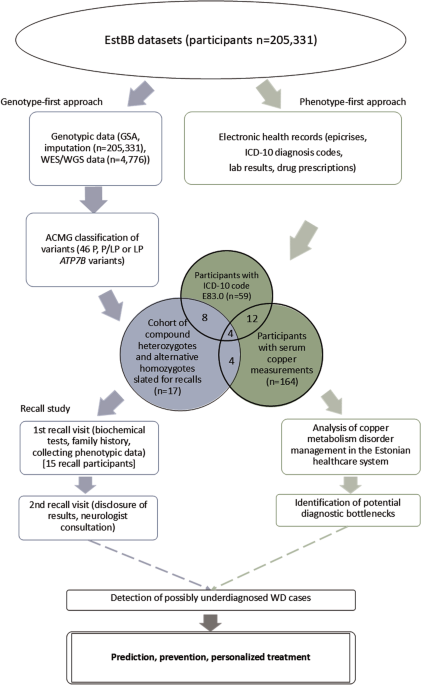
The study employed a combined genotype-phenotype analysis in the EstBB population to investigate WD prevalence and identify potential cases (Fig. 1). The EstBB maintains data, including genotypic and phenotypic data (self-reported medical histories, lifestyle information, EHRs), from 210,000 participants (about 20% of Estonia’s adult population) [14]. Participants provided broad written consent, allowing EstBB to re-contact them and update their data through EHRs and national health registry linkage [15]. As a result, both retro- and anterograde health records are available. This study was conducted using directly genotyped data from Infinium Global Screening Array (v1.0 and 2.0; Illumina Inc., San Diego, CA, USA), enriched by imputed genotype data, available for 205,331 participants, and next-generation sequencing (NGS) data, available for 4776 participants (whole-exome sequencing, n = 2356; whole-genome sequencing, n = 2420). Technical details on direct and imputed genotyping and annotation at EstBB have been provided previously [16, 17]. This study received approval from the Estonian Committee on Bioethics and Human Research.
GSA global screening array (Illumina), ICD-10 International Classification of Diseases, 10th edition; ACMG American College of Medical Genetics, P pathogenic, LP likely pathogenic, EstBB Estonian Biobank, WD Wilson’s disease, WES whole exome sequencing, WGS whole genome sequencing.
Genotype-first approach
Variants in the ATP7B gene and flanking regions (chr13:52499572–52591261, RefSeq build GRCh37) were identified at the EstBB using the high-coverage sequencing, genotyping array, and imputed data. A two-stage filtration process was employed, with consideration of in-silico pathogenicity scores (MetaLR [18], CADD [19], and VEP IMPACT [20]), allele frequencies, and regulatory element overlap, and comparison with clinical variant databases (ClinVar [21] and WilsonGen [22]).
Initially, variants with “moderate” and “high” IMPACT categories were chosen (Supplementary Fig. S1). Variants with “modifier” IMPACT designations and known overlap of regulatory elements (enhancer, promotor, or promotor flanking regions from the Ensembl database [23]) were added to this sample. Variants outside the predefined ATP7B coordinates and those with allele frequencies >1% were excluded. Variants with “pathogenic” (P) and “likely pathogenic” (LP) ClinVar designations were selected. Those without both CADD scores ≥10 and “deleterious” MetaLR predictions were excluded.
In the second filtration stage, variants meeting any of the following criteria were excluded: null-imputation rows (indicating failed variant carrier detection), minor allele frequency (MAF) > 0.0054, and confirmation of benignity in a published study or “benign” or ”likely benign” designation in the WilsonGen or ClinVar database. The filtration was performed in 2020–2021 with the database versions available at the time. Variant classification followed the American College of Medical Genetics/American Molecular Pathologists (ACMG/AMP) 2015 guidelines [24]. The technical details of the follow-up exome sequencing are available in Supplementary Material S1.
Investigating management of copper metabolism disorders in the healthcare system
As part of investigating the management of copper metabolism disorders in the Estonian healthcare system, a phenotype-first approach was used to identify EstBB participants presenting as potential WD cases but without a formal WD diagnosis. The EHR data of EstBB participants who were diagnosed at least once with a copper metabolism disorder [International Classification of Diseases (ICD-10) code E83.0] and who had copper concentrations measured in serum or 24-h urine, were analysed. Epicrises, ICD-10 codes, drug prescription data (Anatomical Therapeutic Classification/Defined Daily Dose codes, dosages, and purchases), and laboratory analyses covering up to 18 years (2004–2023) were retrieved for all study participants. Missing analysis units were inferred from epicrises when available. Individuals with E83.0 diagnoses were categorized as unlikely, possible but unlikely, and possible WD candidates. The criteria for exclusion based on insufficient data included the lack of recurrent E83.0 diagnosis or corresponding epicrisis and the lack of symptoms clearly indicative of WD (presented in epicrises or as ICD-10 codes). To be classified as possible WD, at least two of the following four criteria had to be met: presence of Kayser-Fleischer rings, WD treatment history, low serum copper or ceruloplasmin concentration (or no measurement available), and presentation of overlapping hepatological and neurological/psychiatric symptoms indicative of WD [25]. In unclear cases, a history of alcohol abuse along with the presence of liver damage and the age of first symptom onset (>35 years) were considered as arguments for an “unlikely” classification. EstBB EHRs were further mined for studying the indications behind serum copper testing as part of the management of individuals with copper metabolism disorders. Statistics from the Estonian Health Insurance Fund and Estonian Medicines Agency were explored to compare WD and E83.0 prevalence in Estonia.
Recall study
Genetic findings for EstBB participants identified as alternative homozygotes or compound heterozygotes for P or LP ATP7B gene variants were validated using Sanger sequencing with custom primers at the Estonian Genome Center Core Facility [26]. The recall cohort was chosen based on genetic data after EHR consultation. The study was conducted in 2022–2023. All recall cohort participants received invitation letters with study background information that prompted them to schedule initial recall visits. No personal genetic risk information was disclosed in the letters. Non-responders received a follow-up invitation letter 1 month later, and those who still did not respond were contacted by telephone by biobank personnel.
The recall procedure involved two separate visits. During initial visits, the participants were informed about the process and asked to sign an informed consent form (including consent to the return of results). Their height, weight, blood pressure, pulse rate, and handgrip strength were recorded. Three blood samples collected from each participant were sent to Tartu University Hospital for biochemical analyses, and one sample was used for the secondary confirmation of the genetic findings with Sanger sequencing. The participants were asked about their family and personal medical histories to clarify the findings recorded in their EHRs and preceding events.
During the second recall visits, participants were informed of their genetic findings and counseled on the potential health ramifications, considering their medical histories, lifestyle choices, and biochemical analysis results. An experienced neurologist blinded to the participants’ genetic and medical backgrounds examined them and conducted TCS to detect LN + . The participants were counseled on their examination results and, when necessary, referred for further specialist visits. Detailed information on TCS LN+ and the medical devices used is provided in Supplementary Material S1. Brain MRI data were collected from participants’ EHRs and assessed by the neurologist.
Analysis of p.His1069Gln variant carrier ancestry
Data from individuals with at least one copy of the p.His1069Gln variant, identified from NGS or imputed genotyping data (in 2023), were used to infer: (a) the p.His1069Gln frequency distribution across Estonian counties, (b) patterns of EstBB participant relatedness based on identity-by-descent (IBD) segment sharing, and (c) global ancestry profiles. Allele frequencies were reported by county of birth, disregarding self-reported ethnicity, or by self-reported ethnic group. Confidence intervals for allele frequencies were estimated using binom.test in R (v4.3.0) [27].
IBD segments were detected using IBIS (v1.20.8) [28] with the following calling parameters: -maxDist 0.1 -a 0.00138 -min_l 7 -mt 300 -er 0.004 -2 -min_l2 2 -mt2 150 -er2 0.008. As patterns of relatedness are affected by local demographics [29, 30], the pattern for p.His1069Gln carriers was compared with that for a random sample of EstBB participants matched according to sex, year and county of birth, and self-reported ethnicity. We estimated the average proportions of relatives (individuals sharing at least one IBD segment ≥7 cM) born in each county for the two focal sets, p.His1069Gln carriers and matched participants, and plotted the ratio of these two values on the Estonian map.
The ancestry of each EstBB participant was modeled as a mixture of global ancestry components, as described elsewhere [31]. We focused on Finnish- and Eastern-European–like ancestries, accounting for >90% of the modeled ancestry of 90% of EstBB participants. We grouped p.His1069Gln carriers (n = 2923) and non-carriers (n = 208,337) by the county of birth and divided the median carrier ancestry value by the non-carrier value for each county. Additional technical details are provided in Supplementary Material S1.