
Le 12 janvier dernier, le règlement sur l’évaluation des produits de santé (EU HTA), voté en 2021, entrait en vigueur. Avec quelle conséquence pour une autorité nationale comme la HAS ? “Aujourd’hui, rappelle Lionel Collet, lorsqu’un industriel cherche à se faire rembourser un médicament en France, il s’adresse à la HAS, qui engage un processus en 2 étapes : une évaluation purement scientifique du médicament et l’appréciation, par la Commission de la Transparence, à des fins de remboursement. Une seule de ces deux étapes, la première, devient européenne. La seconde reste à la main des Etats membres. Il y aura donc toujours une Commission de la Transparence, pour donner un SMR (service médical rendu) et une ASMR (amélioration du service médical rendu). “C’est sur la base de l’avis de la Commission de la transparence que le ministère décide d’admettre au remboursement ou non, complète Jean Lessi, et le CEPS (Comité économique des produits de santé) fixe le prix en échangeant avec l’industriel”.
L’architecture de l’EU HTA
Le nouveau règlement européen a fait le choix de mettre en réseau les autorités nationales qui font de l’évaluation des technologies de santé. “Ce choix de ne pas créer de nouvelle autorité centralisée est semblable à ce qui s’est passé dans le champ de la protection des données avec le RGPD”, fait remarquer Jean Lessi. Ce nouveau réseau européen, appelé “groupe de coordination commune” est une assemblée des 27 HAS européennes. Pour chaque médicament ou DM évalué, il va fixer la feuille de route de l’évaluation commune (les fameux PICO, qui définissent la population concernée, les indications, les comparateurs à retenir et les résultats à juger). Dans un deuxième temps, ce groupe des 27 va désigner deux HAS nationales, l’une sera rapporteure, l’autre co-rapporteure. Ce binôme est chargé d’instruire une analyse critique des données dans un projet de rapport conjoint. Enfin, lors de la troisième et dernière phase, le collectif des 27 va adopter (ou discuter et modifier) cette évaluation clinique commune. “Une fois que les 27 HAS ont endossé l’évaluation, elle part dans les 27 États. S’ensuivent les 27 procédures nationales de remboursement et de fixation des prix”, ajoute Jean Lessi.
Ce qui change pour la HAS
Pour Lionel Collet, le nouveau texte permet “une mutualisation de la qualité des évaluations scientifiques, qui vont être homogènes à tous les niveaux. Nous, à la HAS, ajoute-t-il, sommes dans une logique de haute qualité, tous les Etats membres ne l’ont pas au sein de l’UE”. Aussi, cette révolution dans la méthodologie d’évaluation, va dans un premier temps devoir faire l’objet d’une phase de rodage. Le nouveau règlement, ajoute Jean Lessi, va nous amener à travailler avec des autorités qui ont une maturité variable selon les Etats et avec lesquelles nous ne travaillions pas auparavant. Nous allons donc devoir apprendre une culture commune de travail. Les associations de patients, les industriels, les pouvoirs publics nous posent des questions. Nous avons une partie des réponses puisque le Commission européenne a adopté les différents axes d’exécution. Nous allons aussi devoir revoir nos formats de dossiers, nos guides de saisine, mais une partie des réponses sera apportée au fur et à mesure, nous apprendrons en marchant”.
Les réponses aujourd’hui apportées par la HAS nous éclairent sur ce qui change et sur ce qui ne changera pas dans le cadre de l’EU HTA.
Ne changeront pas :
les demandes d’accès précoces post-AMM ;
la procédure de droit commun pour les technologies non concernées par les évaluations cliniques communes (les Joint Clinical Assessments ou JCA) ;
les doctrines des Commissions nationales que sont la CT, le CEESP et la CNEDIMTS, qui continueront de rendre leurs avis selon leurs principes d’évaluation.
Changeront en revanche :
les rencontres précoces nationales : Les médicaments ou dispositifs médicaux ayant fait ou qui feront l’objet d’un JSC ne seront pas éligibles à une rencontre précoce nationale. La priorité sera donnée aux JSC ((Joint Scientific Consultation ou consultation scientifique commune) par rapport aux rencontres précoces nationales. À noter que la procédure de rencontres précoces pour les dispositifs médicaux reste inchangée ;
l’accès précoce post-AMM : “lorsque le médicament a obtenu une AMM, les données soumises au niveau européen ne devront pas être déposées dans tous les dossiers nationaux, précise le règlement (Art 10.3). Il reste cependant possible de déposer des données complémentaires, forcément nouvelles, qui n’auraient pas été versées au dossier initial. “Le règlement européen explique clairement qu’il ne peut pas y avoir deux dossiers, insiste Lionel Collet. Celui de l’évaluation clinique commune “écrase” tous les autres dossiers possibles. C’est donc à nous de voir comment nous allons adapter nos procédures sur ces dossiers d’accès précoce, à partir du travail effectué par le groupe de coordination” ;
les procédures d’évaluation pour le droit commun. Avec la même logique que pour les demandes d’accès précoce post-AMM, les données cliniques fournies au niveau européen ne seront pas redemandées au niveau national. Si de nouvelles données sont disponibles depuis la publication d’un JCA (Joint Clinical Assessment ou évaluation clinique commune), elles devront être déposées dans le dossier national. Si besoin, la HAS peut demander des données complémentaires à l’industriel.
Dispositifs médicaux : ne pas confondre EU HTA et MDR
Interrogés par des industriels du médicament et du dispositif médical le 11 décembre dernier, lors d’un Café Nile, Lionel Collet et Jean Lessi ont dû préciser le cadre d’application du nouveau règlement européen sur l’évaluation des produits de santé, et dire ce qu’il n’était pas. Ainsi, déjà échaudés par les complexités d’application du MDR, les acteurs du dispositif médical craignent de nouveaux retards dans la mise sur le marché de leurs produits. “Le règlement EU HTA et le MDR s’appliquent aux dispositifs médicaux, mais de manière différente. Le règlement MDR s’applique pour une demande d’autorisation de mise sur le marché, en amont de la partie HTA qui concerne une demande de remboursement par l’industriel.
La complexe articulation entre AMM et HTA
Avec le nouveau règlement, l’évaluation des technologies de santé (HTA) va précéder l’obtention de l’AMM, alors qu’aujourd’hui il faut attendre d’avoir l’AMM pour débuter l’évaluation. “Il y aura une parallélisation des procédures, explique Jean Lessi. L’industriel devra déposer sa demande d’évaluation clinique commune au plus tard avant l’avis du comité des médicaments à usage humain (CHMP) et cette évaluation devra être finie au plus tard 30 jours après la délivrance de l’AMM, qui elle même est censée intervenir 67 jours au plus tard après l’avis du CHMP. In fine, il y a donc une fenêtre d’un peu plus de 100 jours pour cet exercice d’évaluation des technologies de santé. Une fois que l’AMM est délivrée, les Etats membres auront donc entre les mains à la fois l’AMM et le rapport d’évaluation clinique commune pour pouvoir rendre leur décision relative à la demande de remboursement.
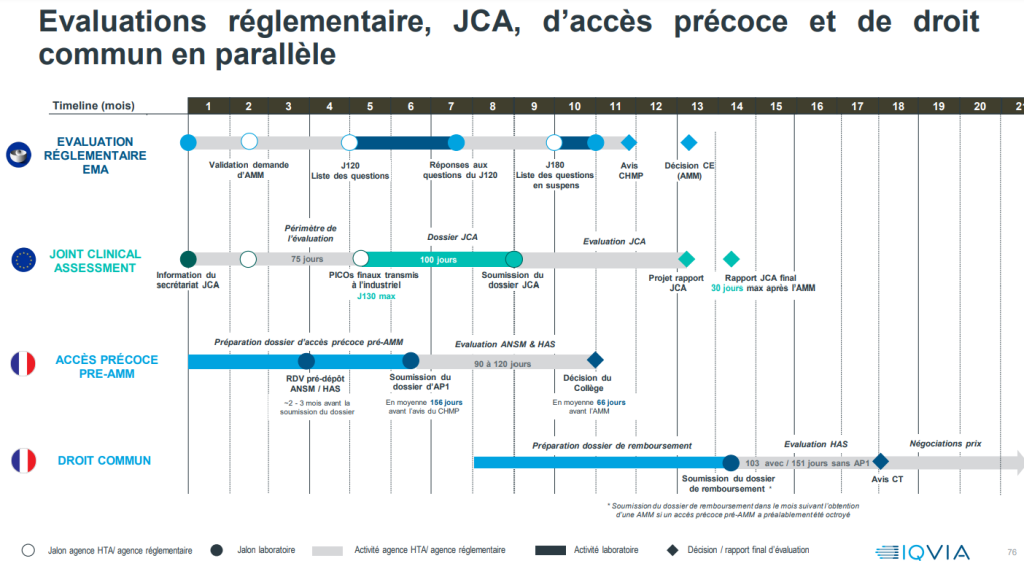
“Les procédures d’AMM et d’évaluation en vue d’un remboursement sont différentes, précise Lionel Collet, même si elles utilisent les mêmes données. La mise sur le marché s’appuie sur un critère de rapport bénéfice/risque absolu. L’évaluation du remboursement intègre une dimension comparative”. À noter que l’articulation entre les deux procédures était jusqu’alors parfaitement séquencée : une fois l’AMM obtenue, l’évaluation à des fins de remboursement peut commencer (exception faite de certaines procédures dérogatoires comme l’accès précoce). Pour le président de la HAS, “il y a une dimension “pari” pris par le législateur européen qui a essayé de viser au plus juste : il s’agit de démarrer à la fois le plus tôt possible, pour ne pas perdre de temps, et le plus tard possible pour avoir des données solides et éviter que les prévisions initiales soient déjouées”.
Lionel Collet s’attend à ce qu’il y ait en 2025 environ 25 nouveaux médicaments à évaluer dans le cadre de l’EU HTA. “La question du DM a encore un peu de temps devant elle, puisqu’elle ne sera pas concernée par le règlement avant 2026”, ajoute-t-il.
Le rôle des associations de patients
Dans le règlement EU HTA, les associations de patients apparaissent dans la première phase d’évaluation (celle de la définition des PICO) comme des interlocuteurs, qui seront consultés. “Les patients doivent être impliqués dans cette phase d’élaboration des PICO dès lors que ce sont les seuls à pouvoir nous parler très clairement du vécu de la pathologie et des aspects relatifs à la qualité de vie, explique Lionel Collet, ajoutant que “au-delà de l’avis du JCA, les patients sont présents au niveau de la Commission de transparence de la HAS.
Aussi, “il est prévu que pour chaque médicament évalué, le groupe de coordination conduise à désigner des experts, y compris des patients (pris individuellement), qui pourront être sollicités par le rapporteur et co-rapporteur. Le règlement prévoit aussi un réseau de parties prenantes où devront figurer notamment des associations de patients transfrontalières, qui pourront intervenir en tant que groupement/personne morale”, précise Jean Lessi.
Les possibilités de recours des industriels
Beaucoup d’ industriels craignent de voir leurs possibilités de recours restreintes avec le nouveau règlement, dans la mesure où ce n’est plus au niveau national, mais bien au niveau européen que se joue l’évaluation clinique. Lionel Collet se veut rassurant et explique que “l’avis rendu est scientifique, donc la question ne devrait pas se poser. Cela signifierait qu’on a une interprétation des données fondamentalement différente de celle de l’industriel. Or, nous travaillerons sur des méthodes rigoureuses”. “Le rapport d’évaluation clinique commune, précise Jean Lessi, n’est d’ailleurs pas, à proprement parler, un avis. Ce rapport rassemble et structure les données scientifiques autour des PICO. L’appréciation dépend, elle, de la Commission de transparence de la HAS. S’il y a débat avec l’industriel, il se fait beaucoup plus sur les phases d’analyse du SMR et de l’ASMR que sur les phases de restitution des données scientifiques collectées”.
Le 12 janvier 2025, le règlement sur l’évaluation des technologies de la santé (ETS ou HTA) est devenu applicable. Objectif : donner un accès plus rapide et plus large aux nouveaux produits innovants et plus efficaces pour les patients. Pour rappel, ces nouvelles règles s’appliqueront d’abord aux demandes d’autorisation de mise sur le marché d’un nouveau médicament anticancéreux ou d’un médicament de thérapie innovante (PTAC). Les règles seront étendues aux médicaments orphelins en janvier 2028 et couvriront tous les nouveaux médicaments à partir de 2030. Certains dispositifs médicaux à haut risque seront également évalués en 2026.
Le 23 janvier 2025, la Commission a adopté les règles relatives aux consultations scientifiques conjointes sur les dispositifs médicaux et les dispositifs médicaux de diagnostic in vitro, dans le cadre du règlement relatif à l’évaluation des technologies de la santé.
Le 3 février 2025, la Commission européenne a lancé la première période de soumission des demandes de consultations scientifiques conjointes (JSC) dans le cadre du règlement sur l’évaluation des technologies de la santé (UE) 2021/2282. Cette période s’étend du 3 février au 3 mars 2025. Pour rappel, les JSC permettent aux développeurs de technologies de la santé (DHT) d’échanger des informations avec les autorités réglementaires sur leurs plans de développement pour un médicament ou un dispositif médical.
La Commission européenne a rappelé que le sous-groupe JCA a effectué six exercices PICO (Population Intervention Comparator Outcome) au printemps 2024 (trois pour les médicaments et trois pour les dispositifs médicaux). Ces exercices ont été réalisés afin de mettre à l’essai et d’améliorer le contenu provisoire des lignes directrices. La Commission les a rendus publiques le 3 février dernier pour des raisons de transparence.