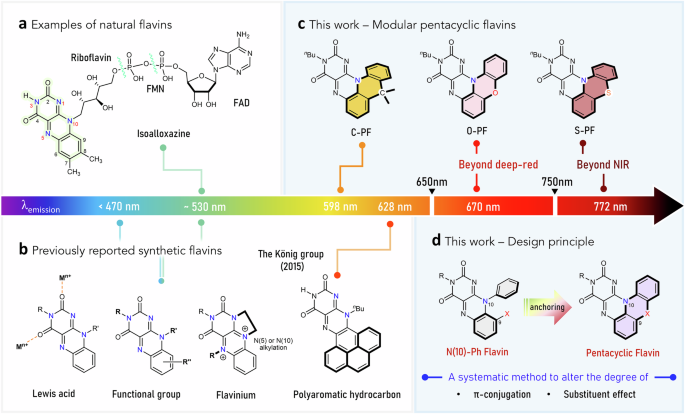
To this end, we designed a pentacyclic scaffold to induce significant changes in the electronic structure, thereby overcoming the intrinsic limits of natural or synthetic flavins in their optical and redox properties (Fig. 1d). Specifically, we hypothesized that anchoring the N(10)-phenyl group to the C(9) atom of the isoalloxazine core via a linker unit (X) would extend π-conjugation and afford a distinctive electronic structure. Furthermore, we envisioned that the identity of X (e.g., O, S, or C(Me)2) would modulate the degree of π-conjugation and introduce specific substituent effects.
In targeting the pentacyclic flavin structure, we initially focused on synthesizing the O-atom-linked variant (Fig. 2a). Nucleophilic aromatic substitution (SNAr) of 2-chloro-1,3-dinitrobenzene with 2-aminophenol, followed by intramolecular SNAr, yielded 1-nitro-10H-phenoxazine. Subsequent hydrogenation produced 10H-phenoxazine-1-amine, which underwent condensation with alloxan to form the pentacyclic scaffold, O-PF-H. For sufficient solubility in organic solvents, we introduced an n-butyl (nBu) substituent at the N(3) position, yielding the desired O-atom linked pentacyclic flavin, O-PF (Fig. 2a). The formation of the targeted five-ring-fused structure was confirmed via multinuclear NMR spectroscopy and high-resolution mass analysis (Supplementary Information).
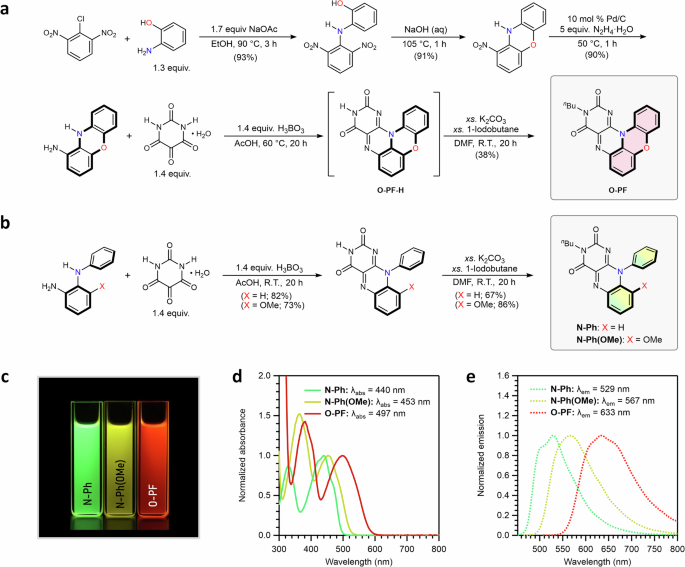
a Synthetic scheme for O-PF. b Synthetic scheme for N-Ph and N-Ph(OMe). c Digital image of THF solutions of N-Ph, N-Ph(OMe), and O-PF taken under 365 nm light. d Normalized absorption spectra of N-Ph, N-Ph(OMe), and O-PF in THF. e Normalized emission spectra of N-Ph, N-Ph(OMe), and O-PF in THF.
Along with O-PF, we prepared two additional flavins as comparative analogs: N-Ph and N-Ph(OMe) (Fig. 2b). In both cases, a phenyl group was substituted at the N(10) atom to investigate the role of the O-atom linker in O-PF from the perspective of extending π-conjugation through ring fusion. In particular, methoxy-appended N-Ph(OMe) was designed to better distinguish the substituent effect of the O-atom from the π-conjugation effect. Similar to O-PF, an n-butyl group was introduced at the N(3) atom in both N-Ph and N-Ph(OMe) to ensure that any observed effects were not influenced by the N(3) substituent.
Remarkably, significant changes in absorption and emission spectra were observed across the series. While the parent compound N-Ph emits typical green light, the pentacyclic O-PF exhibits red emission (Fig. 2c). In THF, the lowest-energy absorption wavelength (λabs) of O-PF is observed at 497 nm, red-shifted by 57 nm compared to that of N-Ph (Fig. 2d). In terms of emission (λem), O-PF shows a maximum at 633 nm, whereas N-Ph exhibits the characteristic green emission of the isoalloxazine core at 529 nm (Fig. 2e). N-Ph(OMe) displays intermediate properties, with λabs at 453 nm and λem at 567 nm, likely due to the electron-donating effect of the methoxy group (Fig. 2d, e). Overall, both absorption and emission wavelengths followed the trend N-Ph N-Ph(OMe) O-PF, reflecting the progressive substituent effect of the O-atom and extension of π-conjugation. The experimental data support our hypothesis that employing the additional π-space and substituent effect into isoalloxazine can be achieved via the pentacyclic platform using a linker unit, such as an oxygen atom.
This significant change in optical properties was further supported by structural analysis. The solid-state structures of N-Ph and N-Ph(OMe), determined by single-crystal X-ray diffraction, revealed that the N(10)-phenyl ring in both compounds is nearly perpendicular to the isoalloxazine ring (Fig. 3a, b). The dihedral angles of C(9a)–N(10)–C(11)–C(16) are 89.2° and 86.7°, for N-Ph and N-Ph(OMe), respectively, indicating an almost orthogonal arrangement between the isoalloxazine and the N(10)-phenyl moieties (Fig. 3a, b).
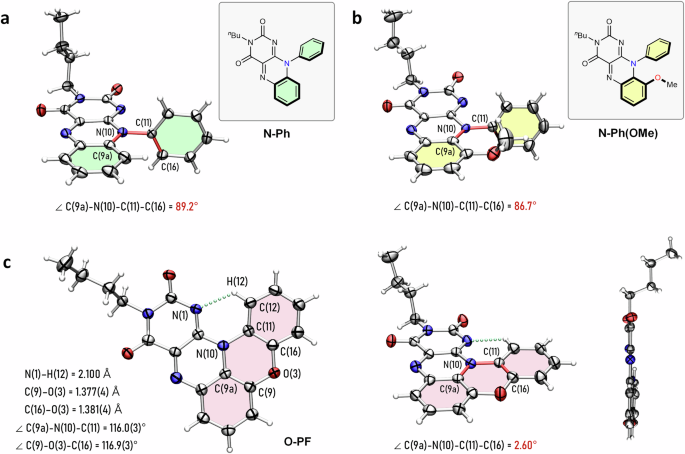
a Solid-state structure of N-Ph. b Solid-state structure of N-Ph(OMe). c Solid-state structure of O-PF from multiple viewpoints. Thermal ellipsoids are set at the 50% probability level (C, gray; N, blue; O, red; H, white). The chemical bonds defining the dihedral angle are colored in red. The dotted line colored in green represents the hydrogen bond.
In stark contrast, the solid-state structure of O-PF revealed a nearly planar geometry (Fig. 3c). The dihedral angle of O-PF is 2.6°, confirming that the fused benzoxazine moiety is nearly coplanar with the isoalloxazine ring (Fig. 3c). Due to this planar conformation, an intramolecular hydrogen bonding interaction, N(1)···H(12)–C(12), was observed in the solid-state structure as revealed by single-crystal X-ray diffraction (Fig. 3c). Selected bond distances and angles within the phenoxazine core, illustrated in Fig. 3c, further corroborate the planarity of O-PF. These findings underscore the pronounced planarity and extended π-conjugation of O-PF.
Of note, in a more polar solvent, such as DMSO, the λem of O-PF was further red-shifted and observed at 670 nm, locating it in the deep-red region. By comparison, the previously reported π-space-extended flavin from the König group emits at 628 nm under similar conditions, establishing O-PF as the most red-emissive synthetic flavin to date (Fig. 1b, c). Although O-PF incorporates fewer additional benzene moieties than the König group’s example (one versus three), it still exhibits a greater red-shift in emission. These results suggest a synergistic performance of the substituent and the π-conjugation extension effects. Therefore, the finding highlights the importance of integrating substituent and π-conjugation effects to modulate the optical properties of flavins.
Synthesis and characterization of C-PF and S-PF
Encouraged by the properties of O-PF, we aimed to expand the emission window into the NIR region by exploring how the identity of the linker unit (X) influences the structural and optical properties of the pentacyclic flavin chromophores. To this end, we synthesized two isoelectronic analogs, C-PF and S-PF.
Akin to the synthesis of O-PF, the condensation of 9,9-dimethyl-9,10-dihydroacridin-4-amine or 10H-phenothiazin-1-amine with alloxan, followed by N(3)-butylation, afforded the five-ring-fused products C-PF and S-PF, respectively (Fig. 4a). Both C-PF and S-PF exhibit significantly distorted geometries in the solid state, as revealed by single-crystal X-ray diffraction analysis (Fig. 4b, c). In C-PF, the dihedral angle is 26.3°, indicating a notable loss of planarity due to the sp³- hybridized carbon at the linker (Fig. 4b). S-PF shows even greater distortion, with a dihedral angle of 36.2°, caused by the larger size of the sulfur atom (Fig. 4c)54. The bulkiness of the sulfur atom significantly alters the bond lengths and angles within the phenothiazine core. Consequently, while O-PF remains nearly planar, C-PF and S-PF adopt a substantially twisted structure to accommodate the sp³-hybridized carbon center or the voluminous sulfur atom, respectively.
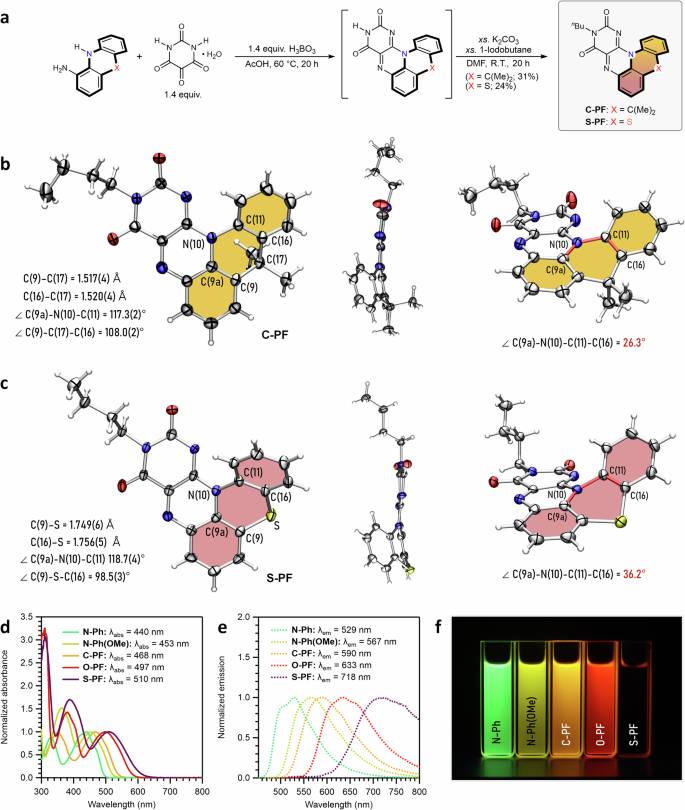
a A summarized synthetic route for C-PF and S-PF. Full synthetic schemes are described in the Supplementary Information. b Solid-state structure of C-PF. c Solid-state structure of S-PF. Thermal ellipsoids are set at the 50% probability level (C, gray; N, blue; O, red; S, yellow; H, white). The chemical bonds defining the dihedral angle are colored in red. d Normalized absorption spectra of the entire series in THF. e Normalized emission spectra of the entire series in THF. f Digital image of THF solutions taken under 365 nm light. The absorption and emission spectra and digital images for N-Ph, N-Ph(OMe), and O-PF are replotted using data in Fig. 2d, e for clearer comparison with C-PF and S-PF.
The perturbed planarity of C-PF and S-PF translated into notable changes in their optical properties. In a THF solution, C-PF exhibited the λabs and λem at 468 nm and 590 nm, respectively—both red-shifted compared to those of N-Ph (Fig. 4d, e). This red-shift is attributed to the extended π-space in C-PF relative to N-Ph, despite its distorted planarity. However, compared with O-PF, the λabs and λem of C-PF remained in a higher energy region, likely due to relatively reduced π-conjugation and the absence of a substituent effect (Fig. 4d, e).
S-PF displayed particularly surprising properties. Although a blue-shift in λabs and λem was expected relative to O-PF, given the bent structure (similar to C-PF)54, S-PF instead showed the most red-shifted λabs (510 nm) and λem (718 nm) in the series. Of note, the λabs of S-PF were red-shifted by 13 nm (512.9 cm–1) compared to O-PF, while its λem was significantly red-shifted by 85 nm (1870 cm–1) (Fig. 4d, e). As a result, the Stokes shift of S-PF was 208 nm (5680 cm–1) in THF, the largest in the series. These results imply that additional factors beyond π-conjugation and substituent effects may contribute to the exceptional Stokes shift observed in S-PF. Overall, both λabs and λem followed the trend: N-Ph N-Ph(OMe) C-PF O-PF S-PF.
Solvatochromism
We hypothesized that the substantial Stokes shift observed in S-PF might result from changes in the dipole moment in the excited state. To investigate this further, we measured the absorption and emission spectra of all five compounds across six different solvents—ranging from non-polar (toluene) to highly polar (DMSO)—to assess solvent effects on their optical properties (Supplementary Information)55.
Figure 5 presents the absorption and emission spectra measured in the least and most polar solvents, toluene and DMSO, respectively. As shown in Fig. 5a, N-Ph displayed nearly solvent-independent absorption and emission properties, along with the smallest Stokes shift (3859 cm⁻¹ in DMSO) within the series. Interestingly, all compounds except N-Ph exhibited solvent-dependent emission. While the absorption spectra showed weak solvent dependence, the emission spectra of N-Ph(OMe), C-PF, O-PF, and S-PF exhibited bathochromic shifts as solvent polarity increased, with the largest Stokes shifts observed in DMSO (Fig. 5b–e and Supplementary Information). Furthermore, each compound showed distinct degrees of solvent dependence, following the order: C-PF N-Ph(OMe) O-PF S-PF (Fig. 5b–e).
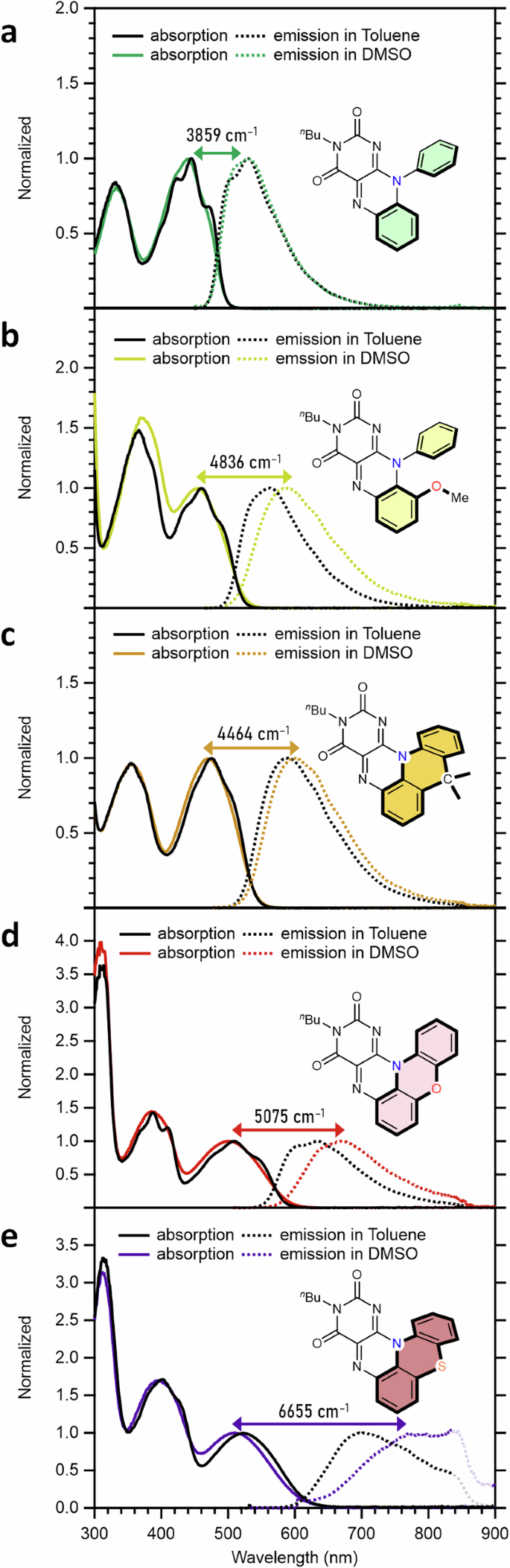
Absorption and emission spectra in toluene and DMSO for a N-Ph. b N-Ph(OMe). c C-PF. d O-PF. e S-PF. Stokes shift values (cm–1) for each compound in DMSO are indicated.
Thus, the Stokes shifts in DMSO are highlighted with double-headed arrows in Fig. 5. As noted, S-PF showed the largest shift, reaching an emission wavelength of 772 nm in DMSO with a Stokes shift of 6655 cm⁻¹ (262 nm). This result marks the example of an NIR-emissive synthetic flavin, highlighting the significant role of the sulfur linker and pentacyclic scaffold in tuning optical properties. Collectively, the experimental data suggest that changes in dipole moment upon electronic transition are enhanced by (1) adopting a pentacyclic structure and/or (2) incorporating a heteroatom unit (e.g., OMe, O, or S).
Computational analysis
Density functional theory (DFT) calculations were conducted to further investigate the electronic structure of the flavin derivatives and address their distinct degrees of solvent-dependent behavior (See Computational Details in the Supplementary Information)56. To optimize computational efficiency, the N(3)-butyl group was truncated to a methyl group. A benchmark study confirmed that the dispersion-corrected B3LYP functional at the Def2-TZVP//Def2-SVP level of theory was the most suitable for describing the spectroscopic properties of O-PF (Table S15)57,58,59,60,61,62. Fig. 6 illustrates the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) of the five flavin compounds synthesized in this study. Notably, the LUMO energy remains largely consistent across the series, with only a subtle deviation from N-Ph to S-PF. Furthermore, the LUMO is primarily localized on the isoalloxazine core in all compounds.
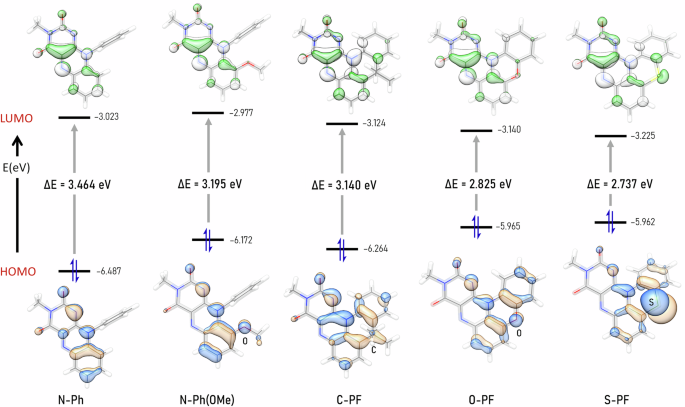
Frontier molecular orbitals and the HOMO-LUMO energy gap (ΔE in eV) of the entire series. Calculated at SMD (DMSO) B3LYP-D3(BJ)/ Def2-TZVP//Def2-SVP level of theory.
In contrast, the HOMO shows distinct variations across the series. In the pentacyclic flavins (C-PF, O-PF, and S-PF), the HOMO is primarily localized on the acridine, phenoxazine, and phenothiazine moieties, rather than on the isoalloxazine core (Fig. 6). This characteristic localization is especially pronounced in O-PF and S-PF, as the lone pairs on the oxygen and sulfur atoms contribute significantly to the HOMO.
The HOMO energy levels of N-Ph(OMe) and C-PF were destabilized by 0.315 eV and 0.223 eV, respectively, relative to that of N-Ph. These changes suggest that both the substituent effect and the additional π-conjugation introduced by ring-fusion contribute to raising the HOMO energy. As such, the HOMO of O-PF and S-PF were further destabilized by the combined influence of these factors. Consequently, the computationally predicted HOMO-LUMO gap decreases progressively from N-Ph (3.464 eV) to S-PF (2.737 eV), showing an overall difference of 0.727 eV. These results align with the experimentally observed trend in λabs values in the absorption spectra of the compounds (Figs. 6 and 4d).
Next, we examined the excited state properties of the flavins to explore the origin of the large Stokes shift observed for S-PF. Time-dependent DFT (TD-DFT) calculations on the S0 → S1 transition reveal that the primary component of the transition is driven by the HOMO → LUMO excitation (Table 1). Additionally, natural transition orbital (NTO) analysis was performed to compute the hole-electron distance (i.e., r(h-e)) for the S0 → S1 transition, which is defined as the distance between the centroids of the NTO hole and NTO electron63,64,65. For N-Ph, r(h-e) is predicted to be approximately 1 Å, indicating a local excitation within the flavin moiety and, thus, a relatively small dipole moment change upon excitation (Table 1). This aligns with the solvent-independent emission behavior observed for N-Ph (Fig. 5a). In contrast, substitution with a methoxy group (e.g., N-Ph(OMe)) or ring fusion into the pentacyclic scaffold (e.g., C-PF) significantly increases r(h-e) to 2.313 Å and 1.740 Å, respectively. These results suggest that N-Ph(OMe) and C-PF experience greater dipole moment changes upon excitation compared to N-Ph.
The enhanced r(h-e) values are consistent with the experimentally observed trends in Stokes shifts and solvatochromism, where a larger dipole moment change upon electronic transition is expected when (1) adopting a pentacyclic structure and/or (2) incorporating a heteroatom unit. Consequently, further enhanced r(h-e) values of 2.403 Å and 2.874 Å were observed for O-PF and S-PF, respectively, as a result of the combined effects of the heteroatom substitution and ring fusion. The localized electron density on the phenoxazine and phenothiazine moieties in the NTO donor (or HOMO) of O-PF and S-PF, respectively, reflects increased charge transfer character upon excitation toward the isoalloxazine-based NTO acceptor (or LUMO).
Overall, the trend in the r(h-e) agrees with the experimentally observed Stokes shifts. For example, N-Ph exhibits neither solvatochromism nor a large Stokes shift, as the electron density rearranges only marginally within the isoalloxazine moiety upon excitation, minimizing changes in the dipole moment. In contrast, for S-PF, excitation induces substantial charge separation between the phenothiazine and isoalloxazine units, leading to a larger dipole moment change and pronounced solvent-dependent emissions with an exceptionally large Stokes shift.
Based on these results, we propose that O-PF and S-PF are capable of emitting deep-red and NIR light, respectively, due to three key factors: (1) extended π-conjugation, (2) substituent effects, and (3) charge separation behavior upon excitation. These factors collectively decrease the HOMO-LUMO energy gap and induce larger Stokes shifts, enabling the development of the deep-red and NIR emissive synthetic flavins.
Electrochemical analysis
The tailored HOMO and LUMO energy levels across the series suggest that the pentacyclic flavin scaffold may exhibit expanded redox capabilities. Upon reduction, each compound displayed a nearly reversible reduction with a peak-to-peak separation around 60 mV (Fig. 7a). For N-Ph, the half-wave potential (E₁/₂) was observed at −1.12 V vs. Fc⁺/Fc, consistent with the one-electron reduction of the isoalloxazine ring5,6,66. Across the series, the E₁/₂ values followed the order: N-Ph(OMe) (−1.13 V) N-Ph (−1.12 V) C-PF (−1.01 V) O-PF (−0.99 V) S-PF (−0.92 V) (Fig. 7a). This trend aligns with the LUMO energy levels predicted by DFT calculations shown in Fig. 6, further validating the agreement between experimental and computational results.
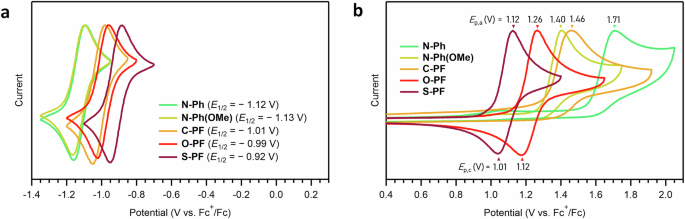
a Reductive scans. b Oxidative scans. Cyclic voltammograms of the entire chemical series obtained in acetonitrile at room temperature using 0.1 M [nBu4N][PF6] electrolyte. Potentials were referenced to the [Cp2Fe]+/0 couple. Scan rate = 100 mV/s.
Unlike reduction processes, oxidation of flavins has received less attention, likely because the isoalloxazine ring is already in its fully oxidized form, analogous to a quinone core. In this work, however, the oxidation is of particular interest due to the significant alterations in the HOMO (Fig. 6). Remarkably, O-PF and S-PF display unusual quasi-reversible oxidation (Fig. 7b). This quasi-reversibility is exclusive to the heteroatom-incorporated pentacyclic flavins (e.g., O-PF and S-PF), as N-Ph, N-Ph(OMe), and C-PF revealed irreversible oxidation (Fig. 7b). The oxidative feature could arise from the formation of stabilized radical cation species, likely due to additional resonance effects within the fused phenoxazine or phenothiazine moieties. Overall, the anodic peak potential (Ep,a) increased in the following order: S-PF (1.12 V) O-PF (1.26 V) N-Ph(OMe) (1.40 V) C-PF (1.46 V) N-Ph (1.71 V) (Fig. 7b). This trend aligns with the HOMO energy levels predicted by DFT calculations (Fig. 6).
These findings provide a strategy for fine-tuning the electronic structure and thus oxidative properties of flavins. Specifically, the developed pentacyclic platform enables selective localization of the HOMO on either phenoxazine or phenothiazine, while preserving the flavin’s characteristic LUMO centered on the isoalloxazine ring. Thus, we believe that this pentacyclic flavin system has the potential to broaden the scope of flavin-mediated redox transformations, and further investigations are ongoing.
In this study, we successfully designed and synthesized a class of flavin-based chromophores, including C-PF, O-PF, and S-PF, to explore how structural modifications and the incorporation of heteroatoms influence their optical and electrochemical properties. Our results demonstrate that altering the nature of the linker unit (X) within the pentacyclic scaffold significantly impacts the π-conjugation, electronic distribution, and redox behavior of these flavins. The developed pentacyclic flavins, O-PF and S-PF, in particular, stand out for their deep-red and NIR emissions, respectively, which arise from a combination of the extended π-space, substituent effects, and enhanced charge separation upon excitation. In addition, the quasi-reversible oxidative feature of O-PF and S-PF demonstrates the potential and capability of this pentacyclic platform to fine-tune the frontier molecular orbitals. These findings not only expand the chemical space of synthetic flavins, marking the examples of deep-red and NIR emissive flavin families, but also open opportunities for their potential applications in bioimaging, photocatalysis, and optoelectronic devices, where tunable redox properties and long-wavelength emission are highly desirable.