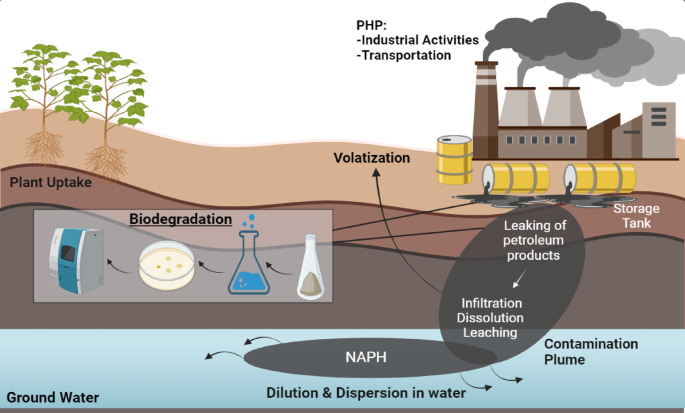
Samples of industrial effluents were collected from Nacharam district, Hyderabad, India (“17.430800 N 78.559500 E”). The selected site encompasses many services and gas stations containing high levels of oil contamination. A soil probe or trowel was used for collecting the soil samples at varying depths (10 cm to 1 m). Many sub-samples were collected, added to a large container, and mixed thoroughly, making it a homogenized mixture. After thorough mixing, a handful of soil (300 mg) was put in 50 mL of pre-sterilized, labelled, and self-sealed Falcon tubes. These sterilized Falcon tubes were then kept at 4 °C in the laboratory and maintained at the same temperature until the next step of analysis21.
Isolation of the microbial consortia
After field sampling, the petroleum-degrading bacteria isolation was carried out according to the method specified in Drifa Yalaoui-Guellal et al. (2017)22. 10 g of the homogenized soil sample was added to a 500 mL flat bottle containing 100 mL of physiological saline (1% NaCl). The mixture was vigorously shaken for 1–2 hours and then allowed to stand for 1 hour. From this mixture 5 mL of supernatant was taken, and subsequently added into the flask containing 45 mL of Mineral Salt Medium (MSM); 1.0 g/L of NH4NO3, 0.7 g/L of MgSO4·7H2O, 3.0 g/L of Na2HPO4, 2.0 g/L of KH2PO4, 3.0 g/L of Na2HPO4, and 1.0 ml of trace element solution. The trace element solution consisted; 0.50 mg/L of CuSO4·5H2O, 30 mg/L of FeCl3, 10 mg/L of ZnSO4·7H2O, 0.50 mg/L of MnSO4·H2O, and 20 mg/L of CaCl2. To promote the growth of the subject bacterial species, 2% (v/v) of crude oil was added to this culture medium and incubated in an orbital shaker at 150 rpm at 30 °C for 7 days. Following incubation, 5 mL of culture was transferred to a fresh medium containing the mixture of MSM medium and crude oil 2% (v/v), and re-incubated at 150 rpm at 30 °C for 7 days.
The experiment was conducted in triplicate. Pure bacterial strains were procured by spreading onto the petroleum-coated MSM agar plates and incubated at 37 °C for 3–4 days. Morphologically distinct bacterial colonies were streaked onto fresh nutrient agar plates and kept at 4 °C for further analysis. The growth curve for the isolated bacteria was obtained by determining the Optical Density (OD) at 610 nm (A610) (Fisher Scientific) for 10–12 days4,23.
Estimation of growth
A 2 mL sample broth was collected after every 24h cycle for 10–12 days. The samples were centrifuged at 4 °C for 15 min at 5000 g. The crude oil extracts were removed by washing the cell pellet with 2 mL of n-hexane and dried at 100 °C in a hot air oven for 24 hours. The bacterial samples were weighed and biomass was determined on a dry weight basis (g/L)24.
Biosurfactant screening of the petroleum degrading bacteria
The selected petroleum-degrading bacteria culture was tested for its potential to produce biosurfactants by undergoing many biosurfactant screening analysis.
Oil – spread method
The oil – spread assay was used to detect the presence of surface-active compounds as described by Rodrigues et al.25, with slight modifications. Briefly, 20 mL of distilled water was added to the petri plates, followed by adding 20 µl of crude oil on the surface of the water. Additionally, 10 µl of cell-free culture broth was introduced to the oil-presented petri plate. After 30 sec–1 min, a clear zone was observed under the visible light. The experiment was conducted in triplicate, and the results were measured in mean ± standard error mean (SEM)26,27,28,29.
Drop collapse test
The drop collapse test was conducted as described by Bodour and Maier30, with slight modifications. The test was employed for detecting the destabilized liquid droplets produced by the biosurfactant. Briefly, the 96-well microplate was coated with 2 µl of crude oil and left to dry for 24 hours. 5 µl of the culture broth was transferred to the oil-coated well plate. The results were determined after a few minutes by calculating the drop sizes. The presence of biosurfactants was observed by a flat drop, whereas the drop remains stable in the absence of biosurfactants. The experiment was conducted in triplicate, and the results were measured in mean ± standard error mean (SEM)31,32.
Hydrocarbon overlay test
This test was primarily employed for identifying hydrocarbon-degrading bacteria and to screen them for biosurfactant production. MSM plates were used containing 40 µl of kerosene, hexadecane, benzene, or toluene as carbon sources. The designated plates were spot-inoculated with the bacterial culture and incubated for 3–7 days at 28 °C. The zones of clearance or halos were observed around the bacterial colonies, which indicated the bacteria’s hydrocarbon degrading potential. The experiment was conducted in triplicate, and the experimental results were represented in mean ± standard error mean (SEM)22,33.
Bacterial adhesion to hydrocarbon (BATH) assay
This method was employed for evaluating the hydrophobic surface characteristics, cell surface hydrophobicity, and measuring the number of bacteria adhering to the hydrocarbon phase. The bacterial cell pellets were collected, washed, and suspended in a buffer salt solution (g/L): 7.3 KH2PO4, 16.9 K2HPO4, and then diluted in the corresponding buffer solution with an optical density of 610 nm (A610). Subsequently, 100 µl of crude oil was added to 2 mL of cell suspension medium and vortexed for 3 mins. After vortexing, the solution was allowed to stand for 1 hour, separating the oil and aqueous layer. OD for the aqueous layer was measured using a spectrophotometer (Fisher Scientific), which calculated the percentage of cells that were adhered to the oil surface34,35. The following formula was used for calculating the percentage adherence:
$$\:\:Bacterial\:Cell\:Adherence\:\left({\%}\right)=\frac{OD\:initial-OD\:shaken\:with\:oil\:}{ODinitial}\times\:100$$
(1)
In which, ODinitial indicated the OD pre-mix value of the cell suspension in the buffer solution, and ODshaken with oil indicated the OD value of vortexed cells using crude oil. In the BATH assay, a few drops of the INT solution (2-(4-iodophenyl)−3-(4-nitrophenyl)−5- phenyl tetrazolium chloride) were added and observed under the light microscope. The viability of the cells attached to the crude oil droplets was indicated by the reduction reaction, which turned the INT solution red. The experiment was conducted in triplicate, and the results were in mean ± standard error mean (SEM)36.
Surface Tension Measurement
The surface tension (ST) reduction of the cell-free culture broth was calibrated using a tensiometer and the Nouy ring method (K20, KRUSS Scientific). The bacterial strains were grown in MSM broth, and 2% (v/v) crude oil was added as a sole carbon source, and the culture was incubated for 7 days at 200 rpm. After incubation, 5 mL of broth supernatant was transferred to a submerged glass tube in a water bath at a constant temperature (28 °C). The height of the ascended liquid in the capillary tube determined the surface tension. The test was performed by taking Triton X-100 solution, 1 mg/mL concentration as a standard control. The experiment was conducted in triplicate, and the results were represented in mean ± standard error mean (SEM)37,38,39.
Determination of critical micelle concentration (CMC)
Different biosurfactant concentrations were prepared in the range 0–100 mg/L using an appropriate buffer solution (Sodium dihydrogen phosphate – disodium hydrogen phosphate of pH 7, 37 °C). The tube that lacked the biosurfactant sample was marked as a negative control. The CMC was demonstrated from the threshold value of the ST versus the logarithm of the biosurfactant concentration curve. The Nouy ring tensiometer (K20, KRUSS Scientific) was employed for determining the ST. The experiment was conducted in triplicate, and the results were represented in mean ± standard error mean (SEM)40,41.
Biochemical characterization
The enumerated bacteria were isolated and stored in a nutrient agar medium at 4 °C for biochemical identification. The primary identification was based on colony formation, cell morphology, and gram staining. The secondary identification was achieved by executing a series of biochemical tests. The observed bacterial structures were compared with Bergey’s Manual of Determinative Bacteriology. All the biochemical tests were performed at 30 °C42,43,44.
Stability test calculation for emulsification index (E24)
The stability test for enunciating the total emulsification index values was conducted according to45,46,47,48,49. For this experiment, 6 mL of crude oil was taken and added to the 4 mL cell-free culture broth, shaken using a vortex for 2 mins, and allowed to stand for 24 hours. The emulsion index (E24) was calculated as a percent ratio of the emulsified zone layer (mm) to the total height (mm), as described in the following equations50,51,52.
$$\:Emulsification\:index\:=\:\frac{Height\:of\:the\:emulsification\:layer}{Total\:height}\times\:\:100\:\left(\%\right)$$
(2)
Media optimization for biosurfactant production
The bacterial culture isolates were grown on MSM medium containing 2% crude oil as the sole carbon source under different physical and nutritional conditions to ascertain suitable parameters for significant hydrocarbon degradation, biosurfactant production, and emulsification index (E24). All the experiments were conducted in triplicate, and results were represented in the form of mean ± standard error mean (SEM).
Effect of pH
To optimize the pH of the solution, 9 pH values (pH: 4–9) were selected. The MSM medium was prepared, and 2% (v/v) crude oil was added as a sole carbon source. The pH of the medium was optimized by adjusting the pH using 0.1 N HCl and 0.1 N NaOH solutions in adequate quantities. After pH adjustment, the media were subjected to sterilization for 15 mins at 121 °C. The activated culture of the isolate was inoculated and incubated at 37 °C in an orbital shaker for 7 days at 150 rpm51,52.
Effect of temperature
To optimize the temperature of the solution, 7 temperature values (10–70 °C) were chosen. The MSM medium was prepared, and 2% (v/v) crude oil was added as a sole carbon source; pH was regulated to 7, and the media were subjected to sterilization for 15 mins at 121 °C. The activated culture of the isolate was inoculated and incubated at chosen temperatures in an orbital shaker for 7 days at 150 rpm51,52.
Effect of carbon
The effect of carbon was determined by taking 8 different carbon sources: diesel, coconut oil, crude oil, maltose, sucrose, starch, glycerol, and mannitol. In the MSM medium, 1% of each carbon source was added, the pH was kept at 7, and then the entire solution was sterilized at 121 °C for 15 mins. The activated culture of the isolate was inoculated and incubated at 37 °C in an orbital shaker for 7 days at 150 rpm51,52.
Effect of nitrogen
The effect of nitrogen was indoctrinated by taking 8 different nitrogen sources: ammonium sulfate, ammonium phosphate, ammonium nitrate, peptone, ammonium chloride, yeast extract, potassium nitrate, and urea. The MSM medium was prepared, and 2% (v/v) crude oil was added as a sole carbon source, and a 1 g/L concentration of each nitrogen source. The pH was adjusted to 7 and the solution was sterilized at 121 °C for 15 mins. The activated culture of the isolate was inoculated and incubated at 37 °C in an orbital shaker for 7 days at 150 rpm51,52.
Effect of carbon and nitrogen concentration
The best-optimized carbon and nitrogen sources were regulated for the best concentrations that were required for maximum yield. The carbon and nitrogen sources were added separately in the MSM medium at different concentrations: 1–7%. The pH of the medium was also adjusted to 7, and then the solution was subjected to sterilization at 121 °C for 15 mins. The activated culture of the isolate was inoculated and incubated at 37 °C in an orbital shaker for 7 days at 150 rpm51,52.
Analysis of the optimized conditions
The bacterial isolates were streaked on fresh agar plates and incubated at 37 °C for 48 hours. The production process was conducted in a shake flask consisting of 250 mL of MSM medium and 2% (v/v) crude oil as the carbon source. 2 mL of pre-cultured bacteria was inoculated into each flask and incubated for 12 days at 28 °C at 200 rpm. Culture media samples were taken and checked at regular intervals to assess the emulsification activity and biomass growth. The proliferation of cells was done by using the dry cell weight technique51,52,53.
Biosurfactant extraction and dry weight calculation
The biosurfactant production was executed in the MSM medium, constituting crude oil as a sole carbon and energy source. The extraction of the biosurfactant was done by centrifuging the fermented broth via a method elaborated in54, with slight modification. Briefly, the subsequent culture of the bacterial isolate was grown on MSM broth overnight. 2% of the pre-bacterial culture was taken and put into the medium exhibiting the biosurfactant production with crude oil, and incubated for 7 days at 25 °C in a shaking incubator at 120 rpm. The solution was subjected to centrifugation for cell removal at 5000 rpm for 20 min at 4 °C. The supernatant was taken, and pH was regulated to 2 by employing 1M H2SO4. Then an equal amount of chloroform: methanol (2:1) was added for biosurfactant extraction, followed by separation of the organic phase and solvent evaporation to concentrate the biosurfactant. The biosurfactant was purified by concentrating the extract in a silica gel (60–120 mesh) glass column and containing chilled acetone as an eluent. The fraction was eluted, and the solvent was evaporated by drying to remove the acetone. Lastly, the purified fraction was dissolved in sterile water, and dry weight was calculated by keeping the dry residues in a sterile petri-plate in a hot air oven for 25 min at 115 °C. After drying, the weight of the plate was determined55. The resulting dry weight of the biosurfactant was calculated by the following formulae:
$$Dry\;weight\;of\;biosurfactant=\left( {Weight\;of\;{\text{ }}plate\;after\,drying – Weight\;of\;{\text{ }}the\;empty\;plate} \right)$$
(3)
All the data were estimated as mean standard error mean (SEM). The experimental data were analyzed by employing the two-way analysis of variance (ANOVA) followed by the Tukey-Kramer multiple comparison tests. The statistical analysis determined that p
Hydrocarbon biodegradation by gravimetric method
The biodegradation profile of the bacterial species was tested by following the methodology by Hossain et al.56, with slight modifications. Briefly, the selected bacterial isolates were inoculated in 100 mL of MSM medium containing 2% crude oil in 250 mL conical flasks on an orbital shaker and incubated at 30 °C at 150 rpm for 21 days. To evaluate the impact of additional carbon sources, a parallel set of experiments was conducted by employing 0.5% glycerol as a supplement to boost biosurfactant production. Control was kept under the same conditions without seeding the bacterial strain.
After incubation, residual crude oil was extracted by using a mixture in a ratio of 1:1 of acetone, and petroleum ether, in which 1 N HCl was added to cease the bacterial activity, and then transferred to a selecting funnel and kept at incubation for 15–20 mins at 120 rpm in an orbital shaker. After incubation, the organic phase was meticulously collected in a pre-weighed beaker, and anhydrous sodium sulfate (Na₂SO₄) was added to remove the moisture content. Subsequently, the solvent was evaporated by employing a rotary evaporator at 50 °C to prevent further thermal degradation of hydrocarbons. The final weight of the beaker exhibiting the residual crude oil was noted, followed by determining the weight of the extracted crude oil by subtracting the initial weight of the empty beaker. The percentage of hydrocarbon degradation was calculated using the following formula57:
$$\:\text{H}\text{y}\text{d}\text{r}\text{o}\text{c}\text{a}\text{r}\text{b}\text{o}\text{n}\:\text{D}\text{e}\text{g}\text{r}\text{a}\text{d}\text{a}\text{t}\text{i}\text{o}\text{n}\:\left({\%}\right)=\frac{\begin{array}{c}Weight\:of\:residual\:\\\:oil\:in\:control\:\:-\:\:Weight\:of\:residual\\\:oil\:in\:isolate\:sample\:\end{array}}{Original\:weight\:of\:the\:residual\:oil}\times\:100$$
(4)
The experiment was conducted in triplicate, and the results were in mean ± standard error mean (SEM). To ensure experimental precision, the pre-weighed beakers were employed in which the complete solvent evaporation was verified before weighing. An abiotic control sample was also included for assessing the non-biological degradation of the crude oil.
Analytical approachPreliminary characterization of the crude biosurfactant
The preliminary characterization was performed by employing two dyes; Sudan Red III (lipid soluble dye), and Methyl orange (water soluble dye) for determining the emulsion by the produced biosurfactants. Further, the total protein and carbohydrate content was estimated using the Folin lowry, and Phenol Sulfuric Acid methods respectively, of the biosurfactant extract58,59.
Nuclear magnetic resonance (1H NMR) analysis
The 1H-NMR spectra were recorded on a Bruker 400 MHz NMR spectrometer (Rheinstetten, Karlsruhe, Germany) equipped with a 5 mm PA BBI 400SI H-BB-D-05 Z probe and using Bruker SampleXpress (BrukerBiospin, Rheinstetten, Germany) for automatic sample change. The sample was dissolved by deuterated chloroform (CDCl3) (99.9%) solvent in a Schotte economic NMR sample tube (5 mm o.d./178 mm length). NMR acquisition and processing were performed as described in Saito et al., 200960 at 25 °C with 5000–5200 accumulations. For each sample, eight scans were recorded with a 90° pulse and a 60-sec relaxation delay with an acquisition time of 2.56 sec to obtain the maximum sensitivity. The start and end points of the integration of the peak area were selected manually. Chemical shifts are given in δ values (ppm) relative to CDCl3 as an internal standard61,62.
Gas chromatographic (GC-MS) analysis
The extracted crude oil samples, both the biodegraded, and abiotic control were tested for fingerprinting, i.e. assessing the molecular, and gross composition by using the gas chromatographic method, as described in Wang et al.63, with slight modifications. The residual alkane fractions were procured at regular time intervals of 10–12 days followed by analysis on a GC-MS gas chromatograph – mass spectrometer (Agilent 7890 A). The extracted alkane fractions were reduced to 200 µL by using a vacuum evaporator. A DMPS capillary column (30 m x 0.25 mm x 0.25 mm) was employed for separating the crude oil components. Helium was used as a carrier gas flowing at a rate of 1.2 mL/min. The column temperature was raised from 80 °C to 240 °C at 5 °C/min and kept at 240 °C for a brief period of 30 mins. The detector and injector temperatures were adjusted to 300 °C and 260 °C respectively. The biodegradation efficiency of the chief hydrocarbons was enunciated by studying the area under the peak in degraded, and abiotic control samples, by following the previously established studies, and the equation below64:
$$Biodegradation\;Efficiency\left(\%\right)=\frac{{{A}_{c}\:-A}_{s\:}}{{A}_{c}}\times\:100$$
(5)
Where Ac, and As are the hydrocarbon degradation efficiency by the abiotic control, and stipulated bacterial sample.
Statistical optimization utilizing experimental design models
Medium components were statistically optimized for procuring the maximum yield, emulsification index of the biosurfactant, and hydrocarbon-degrading potential. It was performed by executing the Response Surface Methodology (RSM) with a Box-Behnken Design (BBD) model. All the experiments were conducted by utilizing the statistical and subsequent experimental design packages in R-programming version 4.2.3.
Box- Behnken experimental design (BBD)
The optimization of the E24 index and hydrocarbon degradation potency was carried out by employing a Box-Behnken experimental design (BBD), a three-level full factorial design (TFFD), and response surface methodology (RSM). These applications were chiefly used to analyze the relationship between the three independent variables and the dependent variable (E24 index and hydrocarbon degradation (%)), and identify the optimal levels of the independent variables for the dependent variable (response). The three independent variables were selected at three different levels: pH (x1), temperature (x2), and salinity (x3) (Table 1). The levels of each variable were determined via a thorough literature survey65,66,67,68. The generalized second-order polynomial model was employed to fit the response surface analysis:
$$\:Y={}_{0}+\:\sum\:_{i=1}{}_{ii}{X}_{i}^{2}\:+\:\sum\:_{i=1}^{n}\sum\:_{j=i+1}^{n}{}_{ij}{X}_{i}{X}_{j}$$
(6)
Where Y is the response variable (E24 index and hydrocarbon degradation (%)), b0, bi, bii, and bij are the regression coefficients for intercept, linear, quadratic, and interaction terms, respectively, and Xi and Xj designate the independent variables (i & j). The models were employed to evaluate the effect of each independent variable on the response69,70,71,72.
Validation of the experimental design
The experiments were set up to validate the factors at optimum conditions of pH, temperature, and salinity suggested by RSM-BBD. The emulsification activity and hydrocarbon degradation potency for the bacterial isolate were determined, followed by some additional tests70,71,72.
Statistical analysis
The effect of independent variables, pH, temperature, and salinity, on the response variables was assessed in triplicate. Analysis of variance (ANOVA) was employed to assess the effects of the designated variables, their interactions, and the statistical significance of the model. The goodness and lack of fit of the 2nd -grade polynomial model equation were expressed by the coefficients of determination R2. The statistical significance and the significance of regression coefficients were also evaluated by F-test at a probability (p) ≤ 0.0570,71,72. The optimal extraction conditions were determined via regression analysis, 2D contour, and 3D response surface plots by using R-programming version 4.3.1.
Isolation and amplification of DNA
After following a series of screening procedures, the genomic DNA of each isolate was obtained from the pure cultures employing the phenol/chloroform extraction method. The purity and integrity of the isolated DNA were analyzed on agarose gel electrophoresis, and the procured bands were amplified using PCR. PCR amplification was carried out for each isolate in a PCR thermocycler exhibiting 50 µL master-mix; 10X Polymerase assay buffer, 1µL of genomic DNA template, 1µL of Taq DNA polymerase enzyme (3U/mL), 4 µL of deoxy-nucleoside triphosphates dNTPs (2.5 mM each), 2µL of universal forward and reverse primers (each), 1X of gel loading buffer, and 30µL of water. Universal primers; 5’- GGATGAGCCCGCGGCCTA − 3’ (forward), and 5’- CGGTGTGTACAAGGCCCGG − 3’ (reverse) were used for the study. Thirty cycles were run to obtain the amplified product, which was further stored at 4 °C. DNA detection was rendered by taking 3µL of each PCR product, which was subsequently loaded onto 2% w/v agarose gel. The gel was stained with 3µL ethidium bromide (10 mg/mL), and subjected to electrophoresis in 1X Tris EDTA Buffer (TEB Buffer (pH 8.3); 89 mM Tris base, 89 mM Boric Acid, 2 mM EDTA dissolved in 1 L of water) for 90 mins at 10 V/cm and 50 mA against the 500 bp ladder as a molecular size standard. The gel components were visualized under a UV transilluminator (Fisher Scientific) or GelDocXR software ( Bio-Rad)73,74,75,76,77.
16S rRNA sequencing
The amplified products were eluted from the gel and purified using a GeneJET Gel Extraction PCR purification kit. Ribotyping was adopted for sequencing the PCR amplified products and performing taxonomic analysis. Owing to the limitations of sequencing, most studies can only use the partial sequences of the 16S rRNA gene product78,79,80,81,82. After attaining the required partial sequences, raw data files of each sample (trace files) were procured and aligned using DNA Baser software v5.15 version. After alignment, the assembled sequences were saved in fasta format and further analyzed using myriad bioinformatics tools and algorithms83,84,85,86,87. The saved fasta files were compared and cross-checked using NCBI – BLASTn, and similar nucleotide sequences were determined88,89.
Evolutionary analysis
The topological similarities between phylogenetic trees were estimated by specific algorithms that took the node structure of a tree into a multi-dimensional model90,91,92. Different algorithms were used to quantify the discrepancies between the sub-trees. The quantification analysis also permitted the exploration of correlative relationships amongst distinct sub-regions of 16S rRNA in terms of phylogenetic resolution. For phylogenetic tree construction, briefly, a taxonomic list was constructed consisting of the obtained sequences from the 16S rRNA sequencing method and the compared sequences from the cross-checked method using BLASTn. The highly similar sequences were aligned using the Multiple Sequence Alignment (MSA) format by utilizing the Muscle algorithm. The Neighbor-Joining (NJ) method from the database set was used for constructing the phylogenetic tree using the Unweighted Pair-Group Method with Arithmetic Averaging (UPGMA), and the Maximum-Likelihood model93,94. The bootstrap test’s relationship with all the taxa was aggregated after 1000 replicates of the tree formation. Molecular Evolutionary Genetics Analysis version XI (MEGA-XI)95,96 software was assigned to conduct the evolutionary analysis.
16S rRNA structure prediction
The secondary structures of 16S rRNA were mainly constructed by employing free energy minimization techniques or algorithms. These free energies were procured from experimental measurements of RNA structural element stability. Since the three-dimensional structure of RNA exhibits a plethora of potential configurations, predicting the equilibrium native structure was an arduous task. Moreover, studies have proven that RNA structures exhibit myriad diacritic motifs, and each motif bears a colossal amount of probable sequence combinations97,98,99. Few of the algorithms, such as the neighbor model, predicted the stabilities of the RNA duplex with only Watson-Crick pair algorithms, proposed by Borer et al., an alternate algorithm proposed by Gray et al.. also explored the thermodynamic parameters for the concerned structure. With the advancement in technology and phylogenetic information, the thermodynamic principles were being articulated for near and good prediction of RNA structure and its properties in duplex formation, along with the determination of spectroscopic properties of RNA100,101,102.
Thus, for determining the secondary structure of the subsequent 16 S rRNA sequences, the Unified Nucleic Acid Folding and Hybridization Package (UNAfold) was employed. The MFold server was utilized by employing the Zuker-Stiegler algorithm for assessing the minimum free-energy structure based on the nearest neighbor model. It further utilized the empirical estimates for determining the thermodynamic properties to demonstrate the secondary RNA structure. Moreover, myriad suboptimal structures were produced, which exhibited a certain percentage of the free-energy ranges. The algorithm further calculated the mole fractions of varied chemical species as a function of temperature, UV absorbance (260 nm), complete melting profiles for melting simulations, heat capacity (Cp), etc.103,104
Microcosm studyPreparation of crude oil polluted soil samples
The microcosms were prepared for determining the bioremediation analysis by the stipulated bacterial isolates, in manually polluted agricultural soil. The composition of the agricultural soil was prepared by following Varjani et al.105, with few modifications. Briefly, 1 kg of pulverized air-dried soil samples was prepared and mixed with 2% crude oil concentration in a large container, followed by five treatment processes, as described in Table 2. Biodegradation was studied under five soil treatment samples; Treatment 1: This lacked all the nutrients, bacteria, or biosurfactant to assess the abiotic loss of crude oil [Abiotic control (AC)], Treatment 2: the nutrients were added but lacked bacteria to assess the biotic loss of crude oil by stimulated microorganisms [Biostimulation (BS)]. Treatment 3: The subsequent bacterial strain SARSHI1 was added but lacked nutrients to assess the effectiveness of the subsequent strains to utilize hydrocarbons [Bioaugmentation (BA)], Treatment 4: The bacterial strains were added along with nutrients for simultaneously assessing the effect [BA + BS]. Treatment 5: nutrients were added along with the biosurfactants, but it lacked the bacterial strain SARSHI1 to assess the role of biosurfactant in enhancing the degradation of indigenous microorganisms [Biostimulation + Biosurfactant (BS + B)]. Treatment 6: In addition to BA + BS, an additional biosurfactant was exogenously added to enhance the bioremediation efficiency [BA + BS + B]. The nutrients were added in a 100:10:1 ratio – Carbon: Nitrogen: Phosphorus (C: N: P), in which glycerol was consumed as a carbon source, NH4NO3 as nitrogen, and Na2HPO4 as a phosphorus source. All this while, the moisture content was maintained at 20% along with the addition of distilled water. Tilling and aeration were done once a week to break the lumps and enhance the homogenization of the soil, which augments the contact between microbes and crude oil106. The experiment was conducted in triplicate, and the results were measured in mean ± standard error mean (SEM).
Phytotoxicity study
The Phytotoxicity Assay was used to determine the biological activity of the stipulated bacterial isolates, following the OECD 2006 guidelines (Organisation for Economic Co-operation and Development OECD, 2006). Two vegetable seeds, Triticum aestivum (wheat) and Vigna radiata (Mung Bean), were selected for the subsequent analysis and primarily treated with sodium hypochlorite. They were then washed with distilled water to remove excess biocide and incubated in the dark for 5–7 days at 20 °C. Petri plates were utilized to evaluate the effect of different treatments (Treatment 1–6) on soil samples. The non-contaminated soil was taken as a positive control. The surface sterilized 10 seeds each were taken in different Petri plates and grown at 25 ± 1 °C with a photoperiod of 16/8 h day/night. Each Petri plate was sprinkled with 30 mL of distilled water daily till the 12thday. Lastly, the germination index was calibrated using Eq. vi107,108. The experiment was conducted in triplicate, and the results were measured in mean ± standard error mean (SEM).
$$\:\text{G}\text{e}\text{r}\text{m}\text{i}\text{n}\text{a}\text{t}\text{i}\text{o}\text{n}\:\text{I}\text{n}\text{d}\text{e}\text{x}\:\left(\text{G}\text{I}\right)\text{\%}=\frac{\varvec{S}\varvec{G}\varvec{e}/\varvec{S}\varvec{G}\varvec{c}}{\varvec{R}\varvec{L}\varvec{e}/\varvec{R}\varvec{L}\varvec{c}}\times\:100$$
(7)
Where SGe and SGc designate seeds germinated in extract and control, while RLe and RLc designate mean root length in extract and control, respectively.