Mouse experiments were approved by and performed in strict accordance with the institutional guidelines of the Institutional Animal Care and Use Committee of Sungkyunkwan University School of Medicine (approval number: SKKUIACUC2021-11-35-1). All animal experiments were performed in a facility approved for BSL-2 experiments involving a bacterial Tn library. Male, 7-week-old BALB/c mice were purchased from Orient Bio (Seongnam, South Korea) and acclimated to an animal facility isolated for infection studies at Sungkyunkwan University School of Medicine. All mice were maintained under pathogen-free conditions on a 12:12 h light:dark cycle, with free access to facility chow and water. The temperature and humidity of the facility were maintained at 22 °C and 50%, respectively. The experiments performed with human blood were all approved by the Sungkyunkwan University Institutional Review Board (IRB: SKKU 2023-01-023).
Cell lines and culture conditions
The lung epithelial cell line A549 (Korean Cell Line Bank, Seoul, South Korea; #10185) was grown in Roswell Park Memorial Institute 1640 (RPMI 1640) medium (Welgene, Gyeongsan, South Korea) supplemented with 10% fetal bovine serum (FBS; Welgene). The murine macrophage cell line Raw264.7 (Korean Cell Line Bank; #40071) and human keratinocyte cell line HaCaT (Cellular Engineering Technologies, Coralville, IA; #CR1017-500) were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM; Welgene) supplemented with 10% FBS. All cell lines were grown in a humidified 37 °C, 5% CO2 incubator and sub-cultured by trypsinization.
Bacterial strains and culture conditions
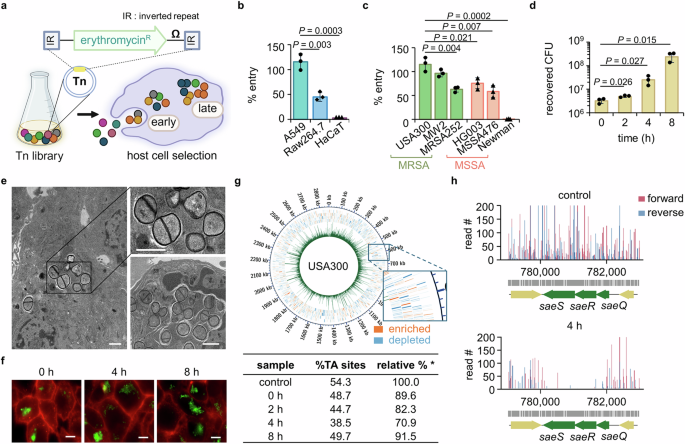
Methicillin-resistant S. aureus (MRSA) strains USA300, MW2, and MRSA252, and methicillin-sensitive S. aureus (MSSA) strains HG003, MSSA476, and Newman strains were grown at 30 °C in liquid tryptic soy broth (TSB; Beckton Dickinson, Franklin Lakes, NJ) with shaking or on agarized TSB plates (TSA). Mutant strains obtained from the Nebraska transposon mutant library (NTML) were grown in liquid TSB or TSA with the addition of 10 µg/mL erythromycin (Erm; Sigma-Aldrich, Burlington, MA). The mutant strains used in the study are summarized in Supplementary Table 1, and all mutants were confirmed for the Tn insertion at the target gene using primers (Macrogen, Seoul, South Korea) as detailed in Supplementary Table 2. On the day of an experiment, an overnight bacterial culture was re-inoculated at OD600 = 0.01 and grown until OD600 = 0.4–0.6. The transposon mutant library (Tn library) was created in USA300 by phage-based transposition as previously described24. Library aliquots were stored at −80 °C and on the day of the experiment were grown from OD600 = 0.2 to OD600 = 0.4 for no more than 3 h.
Generation of mutants of S. aureus
For a generation of ΔmnhF and ΔccpA, DNA fragments corresponding to 1 kb upstream and downstream of mnhF and ccpA were amplified with the primer pairs mnhF1/mnhF3 and mnhF2/mnhF4 for mnhF and ccpA1/ccpA3 and ccpA2/ccpA4 for ccpA (Supplementary Table 2). The kanamycin (Km) resistant gene was amplified with the primer pair KmF/KmR. Following purification, the Km-resistant fragment was combined with the mnhF and ccpA fragments using KOD Xtreme Hot Start DNA polymerase (Novagen, Madison, WI) with the primer pairs mnhF5/KmR and mnhF5/mnhF6 for mnhF and ccpA5/KmR and ccpA5/ccpA6 for ccpA (Supplementary Table 2). The combined PCR product was digested with BamHI/HindIII and BamHI/SaI for mnhF and ccpA, respectively, and the resulting products were cloned into pKFC (pKFC-delmnhF and pKFC-delccpA) and transformed into electrocompetent S. aureus RN4220, resulting in the generation of RN4220-ΔmnhF and RN4220-ΔccpA. Homologous recombination was performed to integrate the plasmid into the bacterial chromosome, and the deletion of the target gene was confirmed by sequencing. Confirmed deletions were transduced using φ11 into HG003 and USA300 to generate HG003-ΔmnhF, HG003-ΔccpA, USA300-ΔccpA50. All bacterial strains, primer sequences, and plasmids are summarized in Supplementary Tables 1 and 2.
Determination and validation of genes essential for intracellular infectionIntracellular infection assay
To determine the extent of intracellular infection and survival of S. aureus, host cells were seeded in the T25 cell culture flasks at 3.5 × 106 cells/flask or the 24-well culture plates at 2.5 × 105 cells/well and placed in a humidified 37 °C, 5% CO2 incubator for 48 h. At 1 h before bacterial infection, the culture medium was replaced with fresh medium. The bacterial strains were grown to a log phase and added to the host cell culture medium at a multiplicity of infection (MOI) of 10 as per the result for screening for MOI that resulted in full recovery of input bacteria. After 1.5 h of infection, bacteria-containing culture medium was removed, and host cells were washed five times with sterilized PBS. Afterwards, medium containing 200 µg/mL gentamicin was added for 1 h to remove extracellular bacteria, after which the host cells were again washed 5 times with sterilized PBS. The host cells were immediately (time point designated as 0 h post-infection throughout the manuscript) lysed with 0.1% Triton X-100, and the collected bacteria were spread on TSA plates and grown at 30 °C for counting of bacterial colony-forming units (CFU). For examination of post-entry events, fresh culture medium was added to the flask after the removal of gentamicin-containing medium and washing with PBS, and host cells were lysed at 2, 4, and 8 h post-addition of fresh medium (time points designated as 2, 4, and 8 h post-infection throughout the manuscript). Bacteria were spread on TSA plates and grown at 30 °C for counting of bacterial CFU. The protocol was adapted and modified from previous publications51,52,53.
Tn library infection experiment
For the determination of genes essential for intracellular entry and post-entry survival, an infection assay of the USA300 Tn library into host A549 cells was performed. Host A549 cells were seeded in T25 cell culture flasks at 3.5 × 106 cells/flask, and the Tn library was grown to OD600 = 0.4 as described above. The infection procedure was performed as described above. The host cells were lysed at 0, 2, 4, and 8 h post-entry and spread on TSA+Erm (10 µg/mL) plates and grown at 30 °C. The colonies were then scraped and pooled for preparation for the transposon sequencing (Tn-seq).
Validation assay with mutants
To validate the results obtained from the Tn library infection assay, individual mutants were tested for the extent of their intracellular entry and post-entry survival54. All mutants are listed in Supplementary Table 1 and were confirmed for the Tn insertion at the target gene using primers as detailed in Supplementary Table 2. For validation, host A549 cells were seeded in 24-well culture plates at 2.5 × 105 cells/well, and the rest of the procedure was the same as outlined above for the intracellular infection assay. Upon lysis of host cells at 0, 2, 4, and 8 h post-entry, mutants were spread on TSA plates containing appropriate antibiotics and counted for the number of colonies at each time point.
Confirmation of the effects of an ATPase inhibitor on bacterial intracellular entry and post-entry survival
For the determination of the effects of ATPase inhibition, USA300 was cultured in media containing 120 µM of oligomycin A (Sigma-Aldrich) for 4 h32. Then, bacteria pre-treated with oligomycin A were utilized for the intracellular infection assay. A549 cells were exposed to bacteria pre-treated with oligomycin A, also in the presence of 120 µM oligomycin A during infection and throughout the experiment until A549 cell lysis at various time points. Upon lysis of host cells at 0, 2, 4, and 8 h post-entry, the bacteria were spread on TSA plates and counted for the number of colonies at each time point.
Determination and validation of genes essential for survival in human blood/plasmaHuman blood/plasma survival assay
To determine the bacterial growth and survival in human blood, human blood anticoagulated with heparin was purchased from Innovative Research (Novi, MI) through an FDA-approved collection center in the USA. To separate plasma, whole blood was centrifuged at 845 × g at 4 °C for 15 min, after which the supernatant was obtained. Whole blood and plasma were placed into 14-mL round-bottom tubes in 2-mL aliquots. The bacterial strains were grown to a log phase and added to the human whole blood or plasma at a final OD600 = 0.02. Whole blood and plasma were incubated at 37 °C, with shaking at 150 rpm for 24 or 36 h. The samples were then placed in red blood cell (RBC) lysis buffer (10×; 1.5 M NH4Cl, 100 mM NaHCO3, 1 mM EDTA, pH 7.4) and spread on TSA plates for CFU counting.
Tn library infection into human blood and plasma
For the determination of genes essential for the survival of USA300 in human blood, whole blood and plasma were prepared as outlined above and placed into 14-mL round-bottom tubes in 4-mL aliquots. The Tn library was grown to OD600 = 0.4 and added to the human whole blood and plasma at a final OD600 = 0.02. Whole blood and plasma were incubated at 37 °C, with shaking at 150 rpm for 24 and 36 h. After 24 and 36 h of growth, samples were placed on ice. RBC lysis buffer was added at a volume of 4 mL and incubated for 10 min to lyse RBC in whole blood samples. Both whole blood (now with lysed RBC) and plasma samples were centrifuged at 9391 × g at 4 °C for 5 min and washed 3 times with sterilized PBS. The resulting pellet was used for preparation for Tn-seq.
Validation assay with mutants
To validate the genes found essential in the blood and plasma survival assay, individual mutants were tested for the extent of their blood survival. All mutant strains are listed in Supplementary Table 1 and were confirmed for the Tn insertion at the target gene using primers as detailed in Supplementary Table 2. For the validation assay, whole blood was placed into 14-mL round-bottom tubes in 2-mL aliquots into which the mutants were inoculated at an initial OD600 = 0.02. The tubes were placed at 37 °C, with shaking at 150 rpm for 24 h after which the bacteria were spread on TSA plates containing appropriate antibiotics and counted for the number of colonies.
Validation of effects of ATPase inhibition on bacterial blood survival
For the determination of the effects of ATPase inhibition, USA300 was cultured in media containing 120 µM of oligomycin A (Sigma-Aldrich) for 4 h. Then, the bacteria pre-treated with oligomycin A were added to human blood at an OD600 = 0.02, also in the presence of 120 µM oligomycin A. After 6, 12, and 24 h of growth, the bacteria were spread on TSA plates and counted for the number of colonies appearing at each time point.
Determination and validation of genes essential for an intra-organ survivalTn library infection into the mouse sepsis model
For the identification of genes essential for bacterial survival in the organs, a mouse sepsis model was used55. Animals were injected with a USA300 Tn library at 2 × 108 CFU per mouse by intravenous injection with an injection volume of 100 μL. For this, the Tn library (grown to OD600 = 0.4 as described above) was resuspended in sterilized phosphate-buffered saline (PBS) used as an injection medium. At 24 h post-bacterial injection, animals were euthanized by isoflurane overdose, followed by exsanguination and organ collection for analysis. At collection, the spleen, brain, heart, liver, lungs, and kidneys were homogenized in PBS containing 0.1% Triton X-100 (Sigma-Aldrich), after which the homogenate was spread on TSA+Erm (10 µg/mL) plates and grown at 30 °C for bacterial colony collection for Tn-seq.
Validation assay with NMTL mutants
To validate the results obtained from the Tn-seq of the organs, individual mutants obtained from the NTML were tested for the extent of their survival in the lungs and liver. All mutants are listed in Supplementary Table 1 and were confirmed for the Tn insertion at the target gene using primers as detailed in Supplementary Table 2. Briefly, mice were injected with individual NTML mutants or wild-type USA300 at 2 × 108 CFU/mouse by intravenous injection with an injection volume of 100 μL. At 24 h post-injection, mice were euthanized, and organs (lungs and liver) were collected and homogenized. The samples were spread on TSA+Erm (10 µg/mL) plates and grown at 30 °C after which the numbers of CFU on the plates were counted.
Tn-seq and data analysis
A Tn library grown on TSA+Erm (10 µg/mL) plates obtained from the intracellular infection study, blood/plasma survival study, and the mouse sepsis study was scraped, and genomic DNA was harvested and prepared for deep sequencing as previously outlined24. Illumina high-throughput sequencing was performed on a Hi-Seq2000 or Hi-Seq2500 for 100 cycles with 40% ΦX174 spiked into the sequencing reaction (Macrogen). The sequencing data were trimmed, filtered, and processed using the Galaxy software suite. The chromosome nucleotide FASTA file for USA300-TCH1516 (NC_010079.1) was downloaded from the NCBI Genome database, and the obtained sequencing data were mapped to the genome using Bowtie software. Python scripts were used to compare the number of reads in the control and experimental conditions using the Mann-Whitney U test, correcting for multiple hypothesis testing with the Benjamini-Hochberg procedure. An EL-ARTIST essential gene analysis was done in MATLAB. The essentiality threshold was set to P value ≤ 0.05 after correcting for the false discovery rate. The operon analysis of the Tn-seq data was performed using Rockhopper (v.2.0.3)56.
Transmission electron microscopy
For transmission electron microscopy (TEM) imaging, A549 cells were seeded in Φ15 mm glass-bottom cell culture dishes (Nest Scientific, Rahway, NJ) at a density of 7 × 105 cells/plate and incubated in a humidified 37 °C, 5% CO2 incubator for 48 h. At 1 h before infection, the cell culture medium was changed to fresh medium, after which USA300 (grown to OD600 = 0.4–0.6) was added to the culture medium at an MOI = 10 for infection lasting 1.5 h, followed by washing (5× with sterilized PBS) and 1 h treatment with gentamicin 200 μg/mL. Cells were then again washed 5× with PBS, and fresh RPMI 1640 medium was added, after which cells were placed in a humidified 37 °C, 5% CO2 incubator for 4 h. At 4 h post-incubation, the sample was fixed in 2.5% glutaraldehyde solution for 1 h at room temperature. The blood samples were generated by culturing USA300 in human whole blood from an initial OD600 = 0.02 for 4 and 12 h at 37 °C, with shaking at 150 rpm. At designated time points, samples were centrifuged at 9391 × g at 4 °C for 5 min and washed with sterilized PBS three times. Then, the samples were fixed in 2.5% glutaraldehyde solution for 1 h at room temperature. Both cell and blood samples were then dehydrated with an ethanol series and infiltrated with Spurr’s resin series, after which the samples were polymerized for 8 h at 60 °C. The sample-embedded blocks were cut with a diamond knife on an ultramicrotome (Leica Microsystems, Wetzlar, Germany), and the resulting sections were directly mounted on 150 mesh copper grids. The sections were stained for 20 min with 2% uranyl acetate in 50% methanol, followed by staining with Reynold’s lead citrate for 10 min. The grids were imaged in a bio-imaging core facility of Korea Institute of Science and Technology (KIST) using Tecnai F20 G2 (FEI, Hillsboro, OR) equipped with a RIO 16 camera under the magnification of 17,000× to 65,000×.
Fluorescence microscopyPreparation of FtsZ-GFP expressing USA300 strain
The pLow-ftsZ-GFP plasmid was inserted into the RN4220 strain by electroporation using a GenePulser cuvette (BioRad, Hercules, CA) at a voltage of 2900 V, 25 μF capacitance, 100 Ω resistance, and 2 mm length57. After electroporation, FtsZ-GFP-expressing RN4220 (RN4220-FtsZ-GFP) colonies were selected on TSA+Erm (10 μg/mL) at 30 °C. The pLow-ftsZ-GFP was then transduced into USA300 using staphylococcal phage ϕ11, generating the FtsZ-GFP-expressing USA300 strain (USA300-FtsZ-GFP). Details of all strains and plasmids can be found in Supplementary Tables 1 and 2.
Fluorescence microscopy
The obtained USA300-FtsZ-GFP was used to visually inspect and analyze bacterial infection into and survival in host cells. For imaging, A549 cells were seeded in Φ15 mm glass-bottom cell culture dishes (Nest Scientific) at 7 × 105 cells/plate and incubated in a humidified 37 °C, 5% CO2 incubator for 48 h. At 1 h before infection, culture medium was changed to fresh medium; and at the time of infection, USA300 (grown to OD600 = 0.4–0.6) was added at an MOI = 10 to the culture medium. After 1.5 h, cells were washed 5× with PBS, followed by exposure to medium containing gentamicin 200 μg/mL. Cells were then washed 5× with sterilized PBS. After being washed with PBS, cells were stained with wheat germ agglutinin Alexa Fluor™ 555 conjugate (Invitrogen, Waltham, MA) for labeling of the cell membrane58. For examination of intracellular survival, fresh RPMI 1640 medium was added, and cells were placed in a humidified 37 °C, 5% CO2 incubator for 4 and 8 h followed by the same staining procedure. All fluorescent images were taken with a Leica DMI8 fluorescence microscope (Leica Microsystems) equipped with a Leica DFC9000 GT VSC-12294 camera under HC PL APO 100× oil immersion lens. Gamma, brightness, and contrast were adjusted identically for the compared image sets using LAS X software (version 3.7.2.22383).
Liquid chromatography mass spectrometryExtraction of blood metabolites
For determination of metabolites in blank plasma, plasma was separated by centrifugation at 845 × g at 4 °C for 15 min, after which 200 μL of plasma was mixed with 400 μL of cold methanol (v/v ratio 1:2). For determination of plasma metabolites upon consumption by bacteria, bacterial culture was added to 2-mL aliquots of whole blood at an initial OD600 = 0.02. The samples were incubated at 37 °C, with shaking at 150 rpm for various time points from 3 to 36 h. Whole blood to which no bacteria were added was used as the control 0 h sample. At the designated time points, whole blood samples were centrifuged for Z.
Extraction of bacterial bacillithiol
For determination of bacterial bacillithiol levels, USA300 was inoculated into whole blood or TSB at an OD600 = 0.02 with shaking at 150 rpm at 37 °C. At 9, 12, and 24 h post-inoculation, samples were centrifuged at 4 °C, 6010 × g for 6 min for the collection of bacterial cells. Bacterial cells collected at a log phase (OD600 = 0.5) were defined as a 0 h sample. The resulting pellet was quenched by the addition of quenching buffer (40:40:20; Acetonitrile:Methanol:Water). Bacterial cells were lysed by mechanical disruption using 0.1 mm glass beads and a precellys homogenizer (Bertin Technologies, Montigny-le-Bretonneux, France) with 12 cycles of 10,000 rpm, 30 sec cycle. Samples were placed on ice in-between each cycle for the prevention of protein degradation. Lysates were centrifuged at 4 °C, 15,871 × g for 5 min, after which supernatant was added to Spin-X centrifuge tubes (Corning Inc.) and centrifuged at 4 °C, 15,871 × g one more time.
Liquid chromatography mass spectrometry analysis
Blood metabolites and bacillithiol were analyzed with time-of-flight (TOF) mass spectrometry (Agilent 6230, Santa Clara, CA) coupled with a liquid chromatography system (Agilent 1290) operated in both positive and negative modes. The mobile phase consisted of solvent A (ddH2O with 0.2% formic acid) and solvent B (acetonitrile with 0.2% formic acid). The gradient condition was as following: 0–2 min, 85% B; 3–5 min, 80% B; 6–7 min, 75% B; 8–9 min, 70% B; 10–11.1 min, 50% B; 11.1–14 min 20% B; 14.1–24 min 5% B followed by a 10 min re-equilibration period at 85% B at a flow rate of 0.4 mL/min. The abundance of extracted metabolite ion intensities was acquired using Profinder 6.059,60. Bacillithiol levels were all normalized by the bacterial CFU measured for each sample.
Growth curve measurement
For the measurement of growth of all mutant strains, strains (grown to OD600 = 0.4–0.6) were added to TSB in 96-well plates at an initial OD600 of 0.002. Growth was observed using a plate reader (BioTek, Winooski, VT) at 30 °C for 20 to 30 h.
Measurement of bacterial minimum inhibitory concentration
To determine the concentration of oligomycin A that does not exhibit any effect on bacteria, a minimum inhibitory concentration (MIC) measurement was performed. Oligomycin A was serially diluted from 120 μM in a 96-well plate in TSB, in which USA300 was grown from OD600 = 0.002 for 24 h. The final OD600 was measured by the plate reader (BioTek) for the determination of oligomycin A concentration affecting bacterial growth.
Cell cytotoxicity
To determine the concentration of oligomycin A which does not exhibit toxicity on host A549 cells, cell cytotoxicity measurement was performed using the CytoTox-Glo™ assay (Promega, Madison, WI), according to the manufacturer’s instructions61. Briefly, A549 cells were seeded in white-bottom 96-well plates at a density of 1 × 104 cells/well. After 24 h of incubation in a 37 °C, 5% CO2 incubator, cells were treated with 0–120 μM oligomycin A for 8 h, after which a substrate that luminesces upon reaction with intracellular organelles but cannot cross the intact membrane of live cells is added to measure cytotoxicity. The fluorescence intensity was measured using the Synergy HTX multi-mode plate reader (BioTek).
Statistical analyses
Statistical analyses between experimental groups were conducted by a two-sided, unpaired Student’s t-test using Microsoft Excel (v.16.98). The results were expressed as the mean ± SD of 3 biological replicates representative of duplicate or triplicate independent experiments. Differences between groups were inferred as significant for P P values are denoted in the Source Data file and the figures.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.