Southern king crab (L. santolla) individuals were obtained from two distinct locations: Ballena Sound and Choiseul Bay, both located in Punta Arenas, Chile. During the sampling period, environmental measurements revealed that Ballena Sound had an average salinity of 24.66 practical salinity units (PSU), reflecting its greater exposure to glacial inputs and associated hydrological fluctuations. In contrast, Choiseul Bay showed slightly elevated salinity at 29.21 PSU. Additionally, Choiseul Bay had comparatively higher temperatures (8.74 °C), dissolved oxygen concentrations (11.17 mg/L), and oxygen saturations (95.36%) than Ballena Sound, which had temperatures of 7.63 °C, dissolved oxygen concentrations of 9.86 mg/L, and oxygen saturations of 92.31%, respectively (Table S2).
De novo gill holo-transcriptome assembly
The sequencing of five biological replicates per location from gill tissue of L. santolla resulted in 10 cDNA libraries. A total of 332,804,332 clean reads were obtained after quality assessment, adapter removal, and rRNA filtering (Table S3). Three holo-transcriptomes are as follows: (i) Global (Ballena + Choiseul), (ii) Ballena, and (iii) Choiseul were assembled to assess microbial composition and host-microbiome functionality between populations (Table 1).
Completeness assessment using BUSCO, based on eukaryotes, arthropods, and prokaryotes orthologs, revealed a high representation of conserved genes. The assemblies shared over 90% of eukaryotic and arthropod orthologs and over 60% of prokaryotic orthologs (Table S4). For the 255 eukaryotic orthologs, the global assembly showed the highest completeness (99.2%), with 66.3% as single copies and 32.9% as duplicates. The Ballena assembly had 96.9% completeness (71.4% single copy, 25.5% duplicates), while Choiseul reached 96.1% (74.5% single copy, 21.6% duplicates). For the 1013 arthropod orthologs, the global assembly showed 94.1% completeness (63.9% single copy, 30.2% duplicates). Choiseul had 92.6% completeness (69.0% single copy, 23.6% duplicates), while Ballena reached 92.5% (70.0% single copy, 22.5% duplicates). Finally, for the 311 prokaryotic orthologs, the global assembly had 65.3% completeness (17.7% single copy, 47.6% duplicates). Choiseul showed 63.4% completeness (17.4% single copy, 46.0% duplicates), and Ballena reached 63.0% (22.8% single copy, 40.2% duplicates).
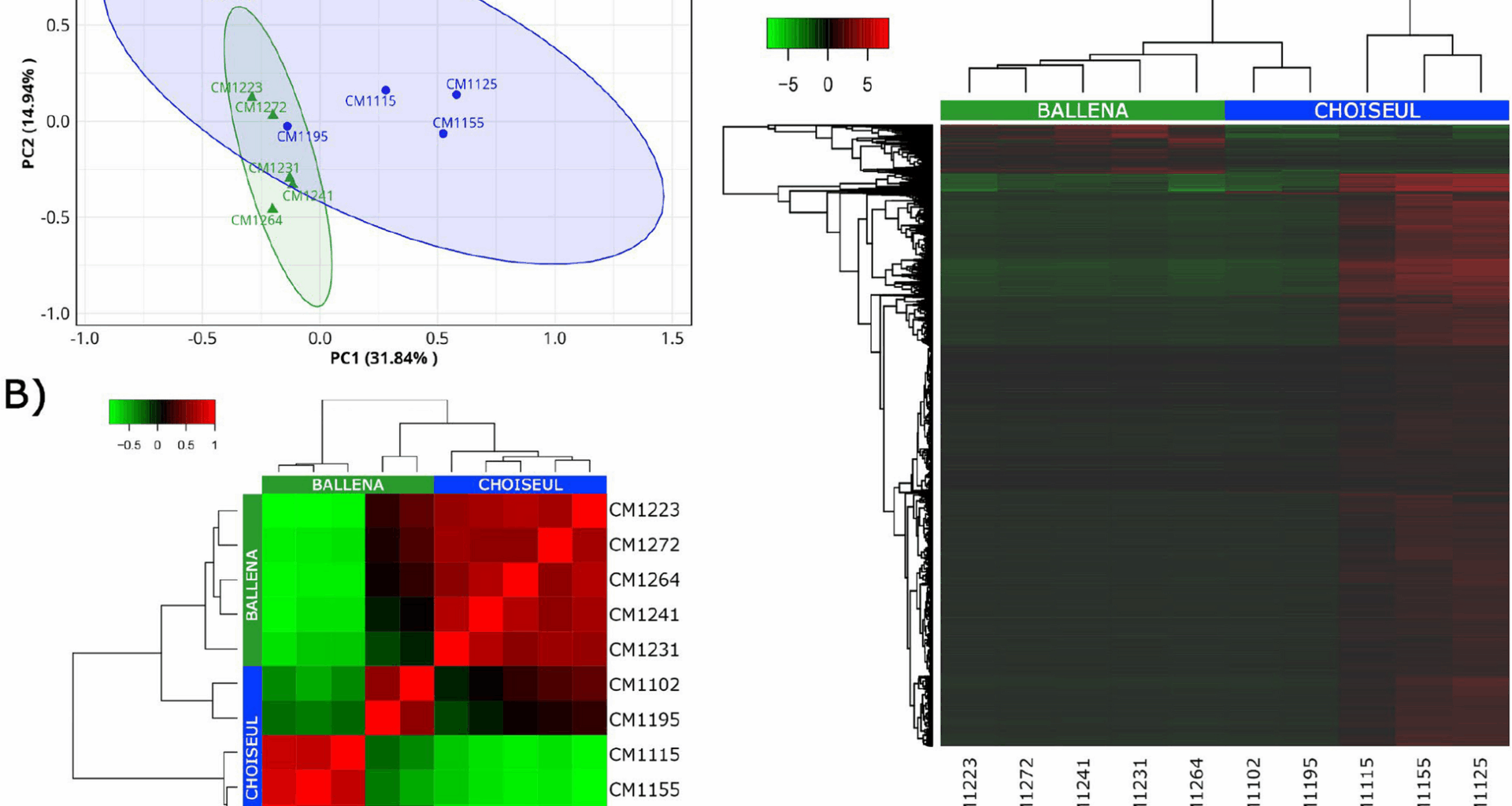
To assess transcriptomic variation across samples, a principal component analysis (PCA) was conducted using RNA-seq data. Principal components PC1 and PC2 accounted for 46.78% of the observed variance (Fig. 1A). However, two samples from Choiseul clustered closer to the Ballena group, suggesting overlapping in their transcriptional profiles. This inter-individual variation was also reflected in the hierarchical clustering, where the same two Choiseul individuals grouped closer to the Ballena cluster (Fig. 1B). Differential expression analysis identified a total of 7819 DEGs between the groups, of which 7207 were upregulated in Choiseul and 612 were upregulated in Ballena (Fig. 1C).
Transcriptomic variation and differential expression patterns between Ballena and Choiseul samples. A PCA plot showing the distribution of samples based on gene expression profiles. Each point represents an individual sample, grouped by locality (Ballena in green triangles, Choiseul in blue dots). Ellipses indicate 95% confidence intervals for each group. B Sample-to-sample correlation heat map based on Pearson’s correlation coefficients. Samples cluster according to their locality, reflecting overall transcriptional similarity within sites. C Hierarchical clustering heat map of DEGs between Ballena and Choiseul. Rows represent DEGs, and columns represent samples. Color gradients indicate relative expression levels (red, upregulated; green, downregulated)
Taxonomic composition of the microbiome in the L. santolla holobiont
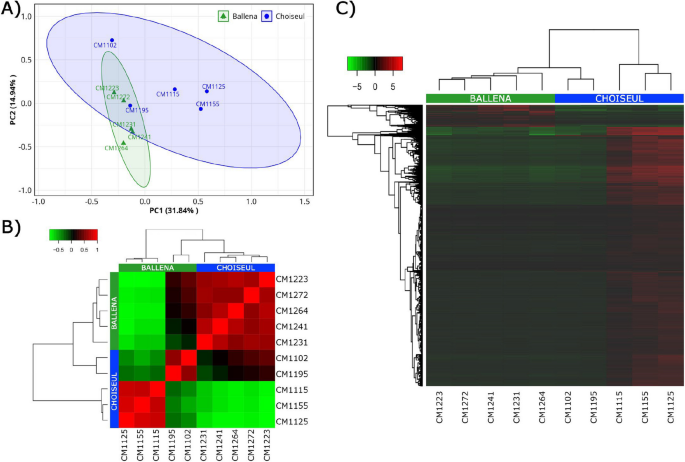
Sequences from de novo holo-transcriptomic assemblies were annotated using the NCBI nonredundant (NR) database. The Ballena assembly contained 68,115 assigned ORFs (40,408 host; 14,565 microbial, and 13,142 other taxa), while Choiseul had 78,513 assigned ORFs (41,369 host; 26,825 microbial, and 10,319 other taxa). Microbial ORFs were classified into archaea, bacteria, fungi, and protists. In Ballena, 32 (0.2%) archaeal, 9763 (67.0%) bacterial, 1594 (11.0%) fungal, and 3176 (21.8%) protist ORFs were identified, while in Choiseul, 56 (0.2%) archaeal, 17,496 (65.2%) bacterial, 1976 (7.4%) fungal, and 7297 (27.2%) protist ORFs were detected.
Archaeal phyla were less represented overall. Thaumarchaeota was the predominant phylum in Ballena, accounting for 0.1% of microbial ORFs (21 ORFs), while in Choiseul, Thermoplasmatota represented 0.06% (17 ORFs) (Fig. 2A). Among bacteria, Proteobacteria was the dominant phylum in both sites, accounting for 43.5% in Ballena (6,336 ORFs) and 40.0% in Choiseul (10,711 ORFs). This was followed by Bacteroidetes, which comprised 12.0% in Ballena (1745 ORFs) and 14.5% in Choiseul (3,890 ORFs), and Verrucomicrobia, representing 2.2% in Ballena (316 ORFs) and 1.8% in Choiseul (490 ORFs) (Fig. 2B).
Relative abundance of microbial phyla associated with the gills of L. santolla from Ballena Sound and Choiseul Bay. Taxonomic profiles are shown for A Archaea, B Bacteria, C Fungi, and D Protists. Bars represent the average relative abundance per site; low-abundance taxa are grouped as “other”
Among fungi, Microsporidia was the most abundant phylum, representing 10.3% of microbial ORFs in Ballena (1505 ORFs) and 6.6% in Choiseul (1766 ORFs) (Fig. 2C). Regarding protists, Ciliophora was the dominant protist phylum, comprising 17.7% of microbial ORFs in Ballena (2572 ORFs) and 18.5% in Choiseul (6314 ORFs). Bacillariophyta accounted for 2.2% in Ballena (317 ORFs) and 1.1% of microbial ORFs in Choiseul (379 ORFs) (Fig. 2D).
Moreover, comparative analysis of microbial composition between Ballena and Choiseul revealed differences in community structure. Bray–Curtis dissimilarity analysis showed a slight difference in microbial community and bacterial composition between conditions (Bray–Curtis = 0.297). Additionally, the chi-square test for taxonomic distribution differences was highly significant (χ2 = 549.60, p
Functional analysis of L. santolla holobiont
Gene Ontology (GO) term analysis was performed to characterize the general functional categories associated with L. santolla and its microbiome from Ballena and Choiseul locations. GO terms were classified into three main categories: Biological process (BP; GO:0008152), cellular component (CC; GO:0005575), and molecular function (MF; GO:0003674) (Fig. S3A). A total of 26,876 genes were annotated from the Ballena assembly, comprising 22,303 host-derived genes and 2290 microbiome-derived genes. In the Choiseul assembly, 31,538 genes were annotated, including 22,711 host-derived genes and 4374 microbiome-derived genes. The number of GO terms per category differed between host and microbiome datasets. In the BP category, the Choiseul host contained the highest number of terms (13,999), followed closely by the Ballena host (13,881) (Table S5).
In contrast, microbiome datasets contained fewer terms—7863 in the CH microbiome and 6175 in the Ballena microbiome. In the CC category, the highest number of terms was recorded for the Choiseul host (1782), followed closely by the Ballena host (1757). Microbiome datasets contained fewer CC terms, with 1127 in the Choiseul microbiome and 913 in the Ballena microbiome. In the MF category, the Choiseul host contained 3611 terms and the Ballena host 3558, while microbiome datasets had 1902 terms in the Choiseul microbiome and 1435 in the Ballena microbiome. A total of 5292 BP terms, 775 CC terms, and 1128 MF terms were shared across all datasets (Fig. S3B, C, D). Overall, host datasets contained the most unique annotated functions, with the Choiseul host exhibiting the highest number of terms across all three GO categories. Likewise, among the microbiome datasets, the Choiseul microbiome showed a higher number of unique GO terms compared to the BA microbiome.
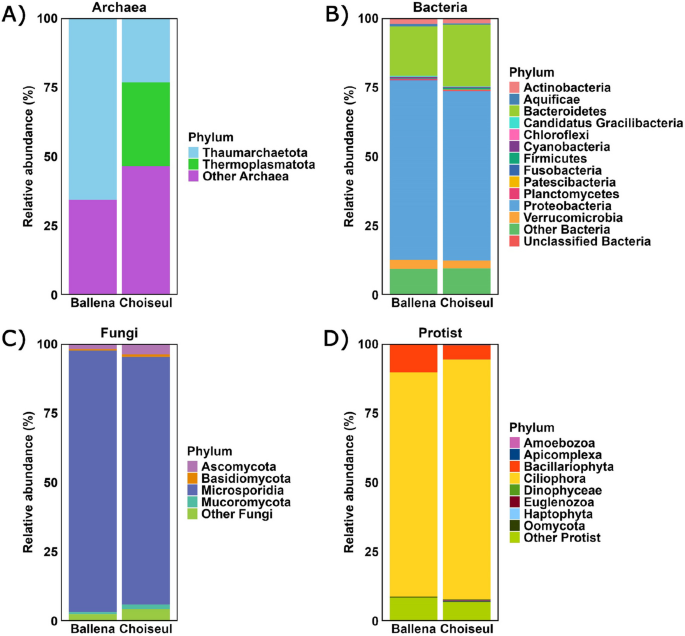
To further characterize the functional profiles of both conditions, enzyme commission (EC) classification analysis was performed. A total of 29,804 proteins were annotated across 3317 EC numbers. Proteins were assigned to six main classes: oxidoreductases (16.0%), transferases (40.6%), hydrolases (28.8%), ligases (5.8%), isomerases (3.7%), and lyases (5.1%) (Fig. 3). Transferases were the most abundant class, with phosphorus-containing group transferases representing 19.1% of all annotated enzymes, primarily in Choiseul host (5.9%) and Ballena host (6.0%). Hydrolases accounted for 28.8% of the dataset, acting on the ester bonds subclass covering 10.0%, most frequently in the Choiseul host (4.3%) and Ballena host (4.2%). Oxidoreductases constituted 16.0% of the dataset, with enzymes acting on the CH-OH group of donors contributing 3.4%, followed by those acting on aldehyde or oxo groups (1.6%). Carbon–oxygen dominated lyases (2.1%), primarily in the Choiseul microbiome (2.5%). Among isomerases, intramolecular oxidoreductases comprised 0.85%, while ligases, mainly involved in carbon–nitrogen bond formation, accounted for 2.0%, with the Choiseul microbiome exhibiting the highest proportion (1.8%). Overall, EC numbers varied across samples, with Choiseul host and microbiome datasets exhibiting a higher repertoire of proteins annotated than Ballena. In addition, the distribution of EC numbers varied across datasets, with the Choiseul microbiome exhibiting the highest proportion of unique ECs (292, 15.1%), followed by the Ballena microbiome (112, 5.8%), Choiseul host (41, 2.1%), and Ballena host (24, 1.2%). Four-hundred thirty-six EC numbers (22.5%) were identified as shared among all datasets (Fig. S4, Table S6). Specifically, the Choiseul host and microbiome shared 4 EC numbers (0.2%), while the Ballena host and microbiome did not share any. Conversely, the microbiome datasets shared 298 EC numbers (15.4%), whereas the host datasets exhibited the highest number of shared ECs, with 493 (25.4%).
Distribution of enzyme classes (EC numbers) in L. santolla and its associated microbiota under Ballena Sound (BA) and Choiseul Bay (CH) conditions. The size of the bubbles corresponds to the number of proteins assigned to each enzyme category, with larger bubbles indicating a higher number of detected proteins
Functional enrichment analysis of DEGs
Functional enrichment analysis was performed to identify differentially enriched GO terms between Ballena and Choiseul comparison (Fig. 4, Table S5). The GO terms were clustered in four main groups, namely membrane transport and ATPase activity, nucleotide metabolism and binding, biosynthesis and macromolecule metabolism, and protein degradation and catabolism. The membrane transport and ATPase activity cluster contained the highest number of enriched terms related to transmembrane transport and ion exchange. The nucleotide metabolism and binding cluster was associated with molecular and ion binding. Biosynthesis and macromolecule metabolism included terms related to protein metabolic processes, whereas protein degradation and catabolism included proteolysis-related processes and metabolic degradation pathways.
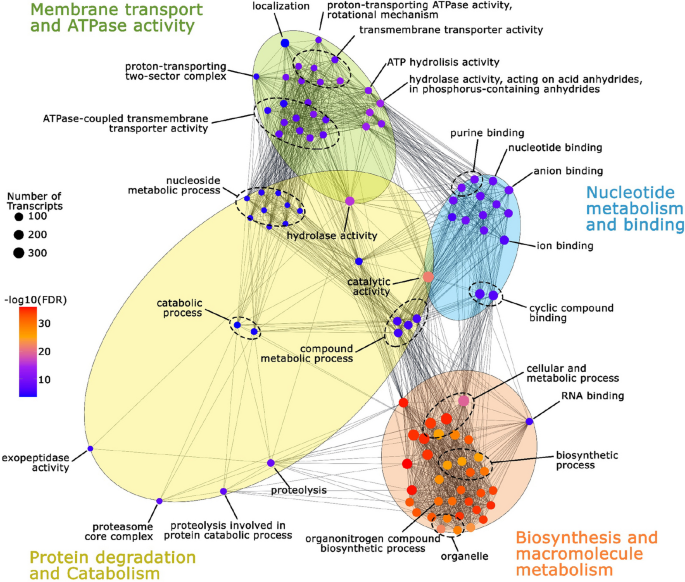
Functional network of enriched Gene Ontology (GO) terms in L. santolla holobiont. The node size corresponds to the number of associated transcripts, while the color gradient (red to blue) indicates the false discovery rate (FDR), with red representing the most significant terms. Functionally related GO terms are grouped into clusters
At the microbiome level, Ballena samples exhibited enrichment in terms such as “carbon fixation” (GO:0015977), which included the upregulated gene RuBisCO (fold change 3.84), while “nickel cation binding” (GO:0016151) featured the nickel-dependent hydrogenase large subunit (fold change 8.77). Other enriched terms included “oxidoreductase activity” (GO:0016491) and “sulfur compound binding” (GO:1901681), with upregulated genes such as ferredoxin oxidoreductase (fold change 6.52) and carbon monoxide dehydrogenase (fold change 5.97). Meanwhile, Choiseul samples exhibited enriched categories such as “active monoatomic ion transmembrane transporter activity” (GO:0022853) and “catalytic activity” (GO:0003824) included the upregulated genes F₀F₁ ATP synthase subunit alpha (fold change 5.28), vacuolar proton-inorganic pyrophosphatase (fold-change 6.18), and Na,H/K antiporter P-type (fold change 6.15). Likewise, “ABC-type transporter activity” (GO:0140359) featured overexpressed genes involved in solute import/export activity.
On the other hand, at the host level, Ballena samples showed enrichment of GO terms such as “enzyme inhibitor/regulator” (GO:0004857/GO:0030234), both of which included the upregulated genes crustin (fold change 2.36) and type IIa crustin (CruIIa-4) (fold change 2.00), as well as “phosphate ion transport” (GO:0006817) and “monoatomic ion transport” (GO:0006811), encompassing upregulated genes such as sodium-dependent phosphate transporter 2 (fold change 2.45) and zinc transporter 2 (fold change 23.0). As for Choiseul samples, enrichment in terms such as “monoatomic cation transmembrane transport” (GO:0098655) and “transmembrane transport” (GO:0055085) was observed, including the upregulated genes ammonium transporter Rh type A (RhAG) (fold change 3.53) and ATP synthase (fold change 5.30). Additionally, enrichment in “antioxidant activity” (GO:0016209), “catalytic activity” (GO:0003824), and other metabolic processes revealed upregulation in genes such as peroxiredoxin (fold change 5.53), thioredoxin-dependent peroxide reductase (fold change 6.04), fumarate hydratase (fold change 7.48), and NADH-ubiquinone oxidoreductase (fold change 4.74).
Enrichment analysis on DEGs revealed enriched KEGG pathways (FDR ≤ 0.05) (Fig. S5, Table S7). At the microbiome level, Ballena samples showed enrichment in metabolic pathways, particularly those associated with carbon metabolism, citrate cycle (TCA cycle), glycolysis/gluconeogenesis, and oxidative phosphorylation. These pathways reflect enhanced energy production and carbon processing functions in this locality. In contrast, Choiseul microbiome samples exhibited enrichment in pathways related to protein turnover and cellular homeostasis, including the proteasome, protein processing in the endoplasmic reticulum, and autophagy. These findings suggest an increased demand for protein folding, degradation, and cellular recycling processes in Choiseul. On the other hand, at the host level, Ballena samples showed enrichment in antigen processing and presentation and endocytosis. In contrast, Choiseul samples showed enrichment in various signaling pathways, proteasome, drug metabolism (other enzymes), phagosome, and endocytosis.