O. boluoensis is a shade-tolerant small tree or shrub. It is distributed at altitudes between 640 and 840 m and generally found along streams. It has leathery leaves, which are simple and alternate (Fig. 1A). Its flowers are white (Fig. 1B), bisexual, mostly in panicles and rarely in racemes. It is outcrossing species and pollinated by insects. In 2018 and 2019, individuals with a diameter at breast height (DBH) larger than 5 cm were observed to flower and set seeds, but only a few actually flowered in these years20. In addition, because it suffers from severe pest/disease attacks on fruit/seeds (Fig. 1C,D), its seed yields were very low. No (healthy) seeds have been collected since 2020. Root-derived clonality was frequently observed in the field (Fig. 1E,F), indicating asexual reproduction. It has green bark (Fig. 1E).

Picture showing Ormosia boluoensis. (A) O. boluoensis leaves; (B) O. boluoensis flowers; (C) and (D) O. boluoensis seeds and fruit showing attack by worms/insects and/or disease; (E) and (F) O. boluoensis growth habitat and its clonal growth with root sprouting.
Chromosome number observation
The individual used for chromosome number observation in O. boluoensis was regenerated from seeds collected from the XTS Reserve. At room temperature, the root tips were pretreated with 0.002 M 8-hydroxyquinoline for 6 h and fixed in 3: 1 (v:v) absolute ethanol:glacial acetic acid for 24 h. The fixed root tips were hydrolyzed in 1:1 (v:v) 1 M absolute ethanol:hydrochloric acid for 7 min, washed by water, and stained with carbol fuchsin for 4 min. Meristems were subsequently excised, squashed and observed by microscope. Photographs were collected using an Olympus BX-43 microscope at 100x magnification with an Olympus DP26 camera. The chromosome number observation in O. boluoensis indicated 2n = 16 (Fig. 2A).
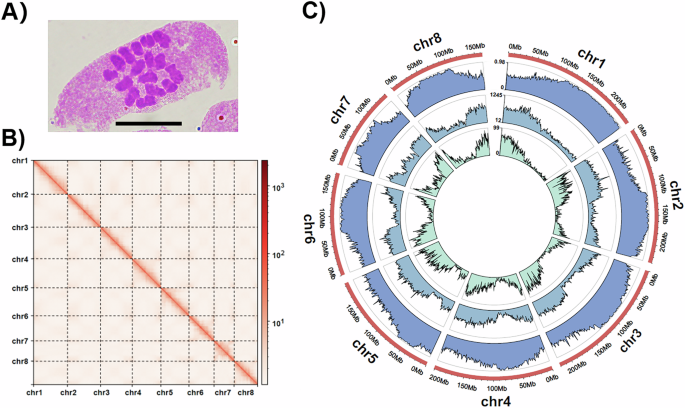
(A) Chromosome numbers observed in Ormosia boluoensis (scale bar: 10 μm); (B) Hi-C interaction heat maps (bin length 100,000 bp) for the O. boluoensis genome assembly; (C) Circos plot showing the genome features (chromosome, repeat density in length proportions, repeat density in numbers, gene density. All densities were estimated with a 1-Mbp sliding window.
Sample collection and sequencing
One O. boluoensis individual collected from the XTS Reserve was used for genome assembly. The collection was approved by the Management Office of Guangdong Xiangtoushan National Natural Reserve under the project of “Research on Manis pentadactyla and the other Rare and Endangered Animals and Plants in Guangdong Xiangtoushan National Nature Reserve — Genetic Diversity and Seedling Breeding of O. boluoensis (2024)”. For genome assembly and annotation, genomic DNA and RNA were isolated from its leaf tissues, and multiple libraries, including long- and short-read whole genome sequencing (WGS), Hi-C and RNA-seq libraries were constructed. Long-read WGS was applied using an Oxford Nanopore Technologies (ONT) PromethION sequencer. Short-read WGS, Hi-C, and RNA sequencing were performed using Illumina HiSeq X Ten platform sequencer with a 150-bp paired-end (insert size 300 bp) sequencing strategy.
Detailed libraries construction and sequencing information, including DNA/RNA preparation and library construction, has been reported in our previous studies21,22. Briefly, genomic DNA of O. boluoensis was extracted using the cetyltrimethylammonium bromide method. Extracted DNAs were used to build short-read and long-read WGS libraries. For the short-read WGS libraries, the DNAs were randomly fragmented to an average size of 200–400 bp for the final library construction. For the long-read WGS library, the extracted DNAs were size-selected and ligated with adapters to construct sequencing libraries. To perform Hi-C scaffolding, leaf tissues of O. boluoensis were immersed into nuclei isolation buffer with 2% formaldehyde for fixation. After fixation, nuclei were sheared into 300–600 bp fragments for the Hi-C library construction. After quality and integrity examination of extracted RNA, the RNA sequencing libraries were generated using RNA library preparing kit. After sequencing, these data were used for genome assembly, annotation, and comparative genomics analysis, with the default parameters in all programs unless otherwise specified.
Finally, the ONT sequencing platform generated approximately 127.13 Gb of WGS reads. The Illumina platform generated approximately 122.07 Gb of short WGS reads, 148.19 Gb of Hi-C reads, and 22.29 Gb of RNA-seq reads (Table 1).
Date pre-processing
After sequencing, the adapters in long WGS reads were trimmed using Porchop v0.2.423 with the parameter “–check_reads 100000 –adapter_threshold 80”. Short WGS and Hi-C reads were quality trimmed using Sickle v1.3324 by removing the reads with base quality values less than 30 and lengths shorter than 80 bp. Short WGS reads were further error corrected using RECKONER v1.125, and duplicates were removed by FastUniq v1.126. Using the error-corrected reads of O. boluoensis, the genome size of O. boluoensis was estimated using Kmergenie v1.704427 with the parameters “–diploid–k 141”. The genome size of O. boluoensis estimated in Kmergenie was 1,467,753,196 bp.
Genome assembly
The O. boluoensis genome was assembled by Nextdenovo 2.3.128 using pre-processed Nanopore long reads with lengths larger than 1000 bp. The specific parameters for our Nanopore reads in Nextdenovo were set using “read_type = ont, input_type = raw” with the others at their default values. After assembly, the genomes were polished by Racon v1.4.2129 and Hapo-G v1.030 using long and short WGS reads, respectively, and each procedure was performed twice. After polishing, Pseudohaploid31 and Purge_Dups v1.2.532 were used to examine and remove duplications in the assembly.
Scaffhic v1.133 was subsequently used to identify misassembly and break the contigs with trimmed Hi-C reads. The Juicer pipeline 1.634 and 3d-dna v20100835 were then applied to perform scaffolding with Hi-C reads. The scaffolding results were visualized using Juicebox v2.04.0636, and the errors were manually corrected when necessary. The major corrections included chromosome boundary adjusting, misjoin and inversion error identifications. After scaffolding, TGS-GapCloser v1.0.137 was used to close the gaps with long WGS reads, and Racon and Hapo-G were used to polish the gap-closed genome. To detect potential chimeric assemblies, we used chimeric-contig-detector v1.0.238 to check them. The program contains two modes of chimera detection: one is “GC-Content Based Detection” mode, and the other is “Read-Pair Based Detection” mode. The latter mode uses short WGS reads.
The initial assembly in Nextdenovo was 1,638,935,015 bp with the contig N50 of 16,933,043 bp (Table 2). The assembly after Hi-C reads scaffolding was 1,565,663,440 bp with the scaffold N50 of 201,058,012 bp, and 1,561,022,207 bp (99.70%) for sequences assembled into 8 chromosomes (Fig. 2B). The largest chromosome size was 236,629,900 bp, and the shortest was 141,093,973 bp. No chimeras were detected under both “GC-Content Based Detection” and “Read-Pair Based Detection” mode by chimeric-contig-detector.
Repeat and gene prediction
Repeat and gene prediction in O. boluoensis followed the procedures previously described by Wang et al.21,22. Briefly, repeats were identified using EDTA v2.0.139 and RED v2.040, and their results were then combined and soft-masked in the assembled genome.
Based on the results of EDTA and RED, 70.35 and 63.04% of genomes, respectively, were identified as repetitive regions. According to EDTA (Table 3), the highest repetitive sequences were long terminal repeats (LTRs), accounting for 61.50% (962,829,486 bp) of the genome size, followed by terminal inverted repeats (TIRs) with 6.74% (105,292,142 bp) of the genome size. For LTRs, the large proportion of repetitive sequences were Gypsy-like, with a sequence size of 555,597,256 bp (35.49%).
Gene prediction was performed using BRAKER3 v.3.0.841 and Funannotate pipeline v1.8.1642. BRAKER3 is a gene annotation pipeline that integrates evidence from transcript reads and homologous proteins. For transcriptome-based gene annotation, RNA-seq reads were aligned to the soft-masked assembled O. boluoensis genome using Hisat2 v2.2.143 and then assembled with StringTie2 v2.2.144. For homology-based gene annotation, protein sequences from 13 reference genomes from Fabaceae (Table 4) were mapped to the assembled genome, generating hints on the gene structure using ProtHint v2.6.045. These results were used to train GeneMark-ETP v.1.046 and AUGUSTUS v3.5.047 to predict genes. The predictions were then combined and filtered by TSEBRA v1.1.2.548 to obtain non-redundant and consensus BRAKER3 gene sets, which were further input into Funannotate to generate the final gene annotation14. After gene prediction, a funannotate pipeline was used for gene functional annotation with different databases, including dbCAN v10.0, EggNOG v5.0.2, Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG), InterPro v5.62–94.0, MEROPS v12.0, Pfam v35.0, SignalP 5.0b and UniProt v2023_0218,19. The reference proteins used for gene predictions are shown in Table 4.
By combining EDTA and RED results, 1,175,171,650 bp (75.06%) of the assembled genome was annotated and masked as repetitive components. The density of repeat sequences in the genome is shown in Fig. 2C. A total of 51,822 genes encoding 56,242 proteins were predicted. Functional annotation of these protein-coding genes showed that 40,750 genes (72.45%) were annotated to at least one database (Table 5).