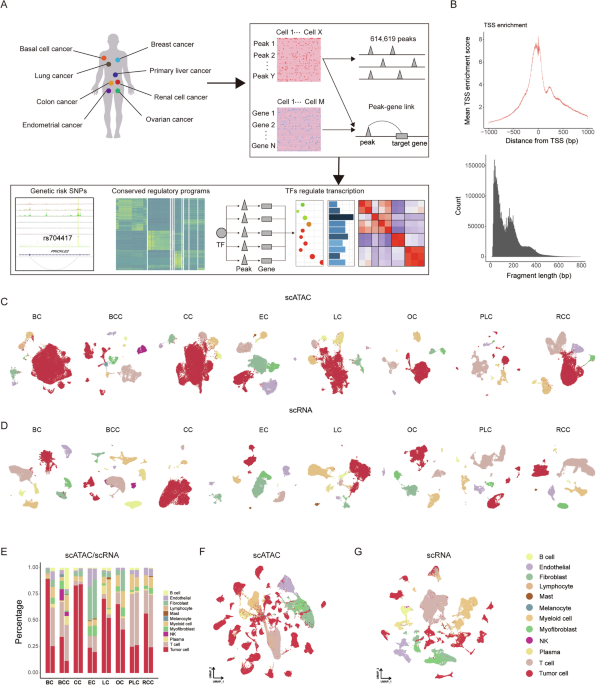
To elucidate the chromatin and transcriptional features of human primary carcinomas, we curated publicly available scATAC-seq and scRNA-seq data, including breast cancer (BC), basal cell cancer (BCC), colon cancer (CC), endometrial cancer (EC), lung cancer (LC), ovarian cancer (OC), primary liver cancer (PLC), and renal cell cancer (RCC) (Fig. 1A, Supplementary Table S1). After rigorous quality control, a total of 176,928 cells from scATAC-seq and 203,537 cells from scRNA-seq were retained for subsequent analysis (Supplementary Table S2). A TSS plot of the data confirms a robust enrichment of unique open chromatin fragments at transcription start sites (TSS), while periodicity in fragment length reveals characteristic peaks indicative of high-quality ATAC-seq experiments (Fig. 1B). Cells were clustered for each cancer type separately and cell type identities were assigned to each cell. To integrate data from different samples, we used the Harmony algorithm [25] for both scATAC-seq and scRNA-seq data to correct batch effects. Cluster annotations were accomplished by comparing differentially accessible regions and differential gene expression with markers associated with various cell types, including tumor cells (LGR5, EPCAM, CA9, KRT18), T cells (CD247), NK cells (NCR1), plasma cells (JCHAIN, FCRL5), myofibroblasts (ACTA2), myeloid cells (ITGAX, CD163), mast cells (KIT), fibroblasts (PDGFRA), endothelial cells (EMCN, PECAM1, PLVAP), melanocytes (MLANA), and B cells (MS4A1) (Fig. 1C, D, Supplementary Fig. S1A, B, Table S3). At the same time, we identified primary copy number variation (CNV) events in each cell based on its transcriptomic profile to confirm the annotations of tumor cells by marker genes (Supplementary Fig. S1C). We presented the cell composition proportions for both scATAC-seq and scRNA-seq datasets in all cancer types (Fig. 1E). Moreover, we projected all the cells onto a UMAP, and cells identified as the same type clustered as expected (Fig. 1F, G).
A Schematic representation of the cancers used in this study, and downstream bioinformatic analyses. B Enrichment of scATAC-seq accessibility near transcription start sites (TSS) and fragment length distribution across all datasets. C UMAP plots of scATAC-seq cells color-coded by cell types found in eight carcinomas. D UMAP plots of scRNA-seq cells color-coded by cell types found in eight carcinomas. E Proportion of various cell types in scATAC-seq (left) and scRNA-seq (right) for eight types of carcinomas. UMAP plots show the embedding of integrated scATAC-seq data F and scRNA-seq data G.
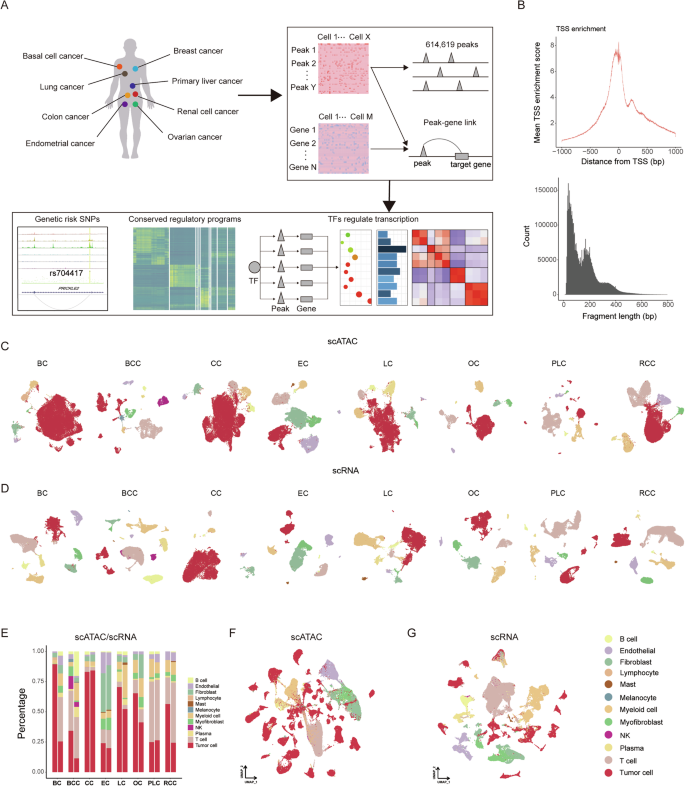
To identify variances in chromatin accessibility across diverse cell types, we identified 257,632 peaks in BC scATAC-seq data, 138,534 peaks in BCC scATAC-seq data, 220,003 peaks in CC scATAC-seq data, 329,815 peaks in EC scATAC-seq data, 263,338 peaks in LC scATAC-seq data, 326,761 peaks in OC scATAC-seq data, 162,992 peaks in PLC scATAC-seq data, and 167,873 peaks in RCC scATAC-seq data individually, utilizing MACS2 (Fig. 2A). Most of them were observed in more than 10 cells (Supplementary Fig. S2A). The predominant presence of these accessible peaks is noted in intronic, distal intergenic, as well as promoter regions of the genome. Intriguingly, our scATAC-seq approach detects more open chromatin regions than those identified by bulk-tissue ATAC-seq (Fig. 2B). We then merged the accessible chromatin regions to obtain a list of 614,619 non-overlapping peaks, which covered 61.5% of the regulatory elements in the registry of cCREs published by the ENCODE consortium [50] (Fig. 2B), suggesting that scATAC-seq extends the compendium of DNA regulatory elements. In accordance with published findings [3, 51], which establish an inverse correlation between DNA methylation and chromatin accessibility at regulatory elements, we examined the methylation data from TCGA to further delineate the accessible potential of the peaks identified. Notably, methylation sites within peak regions exhibit lower methylation β values, while those in non-peak regions display higher methylation β values (Supplementary Fig. S2B).
A Quantification of peaks identified in individual cancer-type scATAC-seq datasets and the combined dataset. Colors correspond to the indicated peak types. B Venn diagram illustrating the intersection between cCREs identified in scATAC-seq data and the registry of cCREs created by other projects. This includes BC scATAC-seq data intersecting with TCGA-BRCA bulk ATAC-seq data, CC scATAC-seq data intersecting with TCGA-COAD bulk ATAC-seq data, EC scATAC-seq data intersecting with TCGA-UCEC bulk ATAC-seq data, LC scATAC-seq data intersecting with TCGA-LUAD bulk ATAC-seq data, PLC scATAC-seq data intersecting with TCGA-LIHC bulk ATAC-seq data, RCC scATAC-seq data intersecting with TCGA-KIRC bulk ATAC-seq data, and integrated scATAC-seq data intersecting with the ENCODE consortium. The numbers on the left, right, top and bottom represent the number of unique peaks in scATAC-seq data, the number of unique peaks in TCGA data, the number of overlapping peaks in scATAC-seq data, and the number of overlapping peaks in TCGA data, respectively. C The number of DARs for all cell types in the scATAC-seq data of eight carcinomas. D Normalized scATAC-seq tracks for all cell types in CC (upper) and the eight types of tumor cells (lower) at the SNP rs4554811 locus. E Normalized scATAC-seq tracks for all cell types in CC (upper) and the five types of fibroblasts (lower) at the SNP rs704417 locus. Dot plot showing the significance of enrichment for selected traits from BC F, CC G, and OC H, within DARs from different cell types. Each circle represents a cell type.
We then identified differentially accessible chromatin regions (DARs) across distinct cell types. Tumor cells, endothelial cells, and fibroblasts showed a higher prevalence of DARs, whereas myeloid cells, plasma cells, and B cells demonstrated comparatively fewer DARs (Fig. 2C, Supplementary Table S4). To examine the heterogeneity within the same cell type across different cancers, we identified cell-type-specific cancer-associated DARs. These analyses indicate that tumor cells exhibit the highest degree of heterogeneity across the eight types of cancer, followed by fibroblasts, while immune cells display minimal heterogeneity (Fig. S2C, Supplementary Table S4).
Single-cell ATAC-seq can reveal distinct cancer genetic risks
To determine the impact of our identified accessible chromatin regions on the genetic variation associated with carcinomas, we scrutinized the NHGRI-EBI GWAS catalog for BC, CC, EC, LC, OC, PLC, and RCC, and noted a higher prevalence of SNPs within our identified accessible chromatin regions in BC, CC, and OC (Supplementary Fig. S2D). By overlaying the co-accessibility map with the chromatin accessibility signal and GWAS statistics along the genomic axis, we were able to identify known chromatin accessibility sites. SNP rs4554811 and rs704417 are associated with increased susceptibility to colon adenocarcinoma, consistent with the presence of focal chromatin accessibility in CC scATAC-seq data [52, 53]. SNP rs4554811 located intronically in VTI1A, exhibits heightened accessibility in colon tumor cells, with weak accessibility in other tumor cells, presenting its specific inherited risk in CC susceptibility (Fig. 2D). Additionally, SNP rs704417 shows strong accessibility in fibroblasts from CC, as well as from EC, OC, LC, and BC, suggesting a potential role for it in previously unappreciated cancer contexts (Fig. 2E). Further analysis revealed that numerous GWAS SNPs were open in multiple cancer types, suggesting that they play similar regulatory roles across different cancer types (Supplementary Table S5).
To further investigate the genetic risk signals for BC, CC, and OC, we performed cell-type-specific linkage-disequilibrium score regression (LDSC) analysis of our scATAC-seq clusters using GWAS summary statistics in BC, CC, OC, as well as other relevant traits (Fig. 2F–H, Supplementary Table S6). Intriguingly, immune-related diseases, such as systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), ulcerative colitis (UC), and Crohn’s disease (CD), exhibit a notable correlation with immune cells. Additionally, fibroblasts and myofibroblasts exhibit associations with traits such as balding, coronary atherosclerosis (CAD), and body height (BH), consistent with prior investigations [54]. With respect to BC, GWAS SNPs were enriched in open regions from tumor cells, and in CC, both fibroblasts and tumor cells display a significant enrichment for GWAS SNPs, whereas in OC, enrichment was observed in fibroblasts and myofibroblasts. We further revealed that the expression of marker genes in CC fibroblasts, OC fibroblasts, and OC myofibroblasts presented significant correlations with overall survival, suggesting dysfunctions in these stromal cells contribute to a worse prognosis (Supplementary Fig. S3A–D).
Differentially accessible regions are candidate DNA regulatory elements
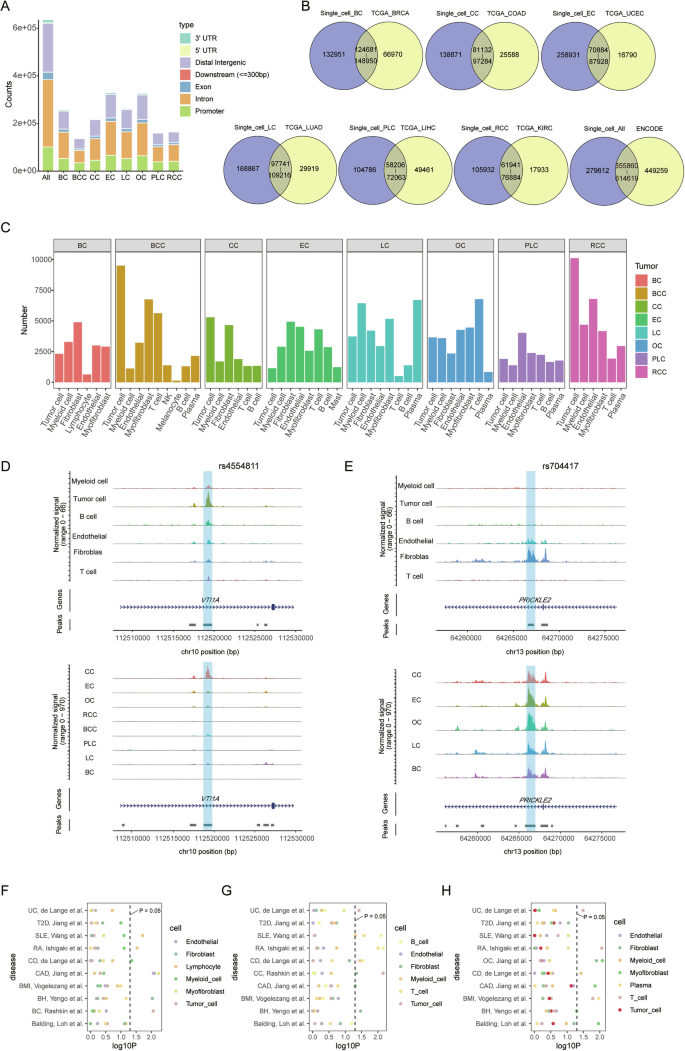
To investigate potential variations in regulatory dynamics across different cancer types, we visually examined our scATAC-seq data. The regulatory elements of MYC, known for their high degree of tissue specificity and distinct locations in different cancer types [55], served as our focal point. Our analysis revealed that MYC is widely expressed and its locus has extensive accessibility in various cell types from the tumor ecosystem (Supplementary Fig. S4A, B). Remarkably, tumor cells exhibit extensive chromatin accessibility at both the 5′ and 3′ DNA elements of MYC, whereas other cells primarily display accessibility at the 3′ DNA elements. Furthermore, we focused on tumor cells extracted from integrated scATAC-seq data (Supplementary Fig. S4C, D). Our finding revealed that tumor cells from BC, BCC, CC, EC, LC, and OC all have strong accessibility at both the 5′ and 3′ DNA elements of MYC, while PLC and RCC show accessibility at the 3′ regulatory elements, consistent with previous findings [3] (Fig. 3A). The comprehensive nature of our scATAC-seq data is underscored by its ability to reveal the enhancer regions of MYC in CC, RCC, LC, and OC, reinforcing its credibility (Fig. 3A). We next turned our attention to NDGR1, a gene known for its pivotal role in cancer development and progression, but for which a comprehensive understanding of its transcriptional regulation is lacking. Notably, the top 5 regulatory elements documented in GeneCards surface in our scATAC-seq data analysis (Fig. 3B). Differential accessibility of these regulatory elements across distinct tumor cells suggests varied regulatory influences. Additionally, we identified a region (chr8-133320109-133321082) exhibiting high accessibility in BCC, EC, OC, and RCC, which is conspicuously absent in CC cells (Fig. 3B, C).
A Normalized scATAC-seq tracks for the eight types of tumor cells at the MYC locus. Enhancer regions of MYC are colored in yellow. B Normalized scATAC-seq tracks for the eight types of tumor cells at the NDRG1 locus. Reported enhancer regions of NDRG1 are shaded in yellow and the predicted enhancer region is marked in gray. C The rank of DARs for colon cancer cells with low accessibility. D Workflow of the construction for peak-gene link network. The triangle represents cells derived from scATAC-seq data, while the circle represents cells from scRNA-seq data. Different colors indicate distinct cell types. E Connection plots illustrating the interactions between the promoter region and predicted enhancer region for NDRG1 in BC, BCC, EC, LC, OC, PLC, and RCC. The p-value of peak-gene represents the p-value from the peak-gene correlation analysis. The co-accessibility score of peak-promoter represents the co-accessibility score calculated by Cicero.
To explore the interaction between DNA regulatory elements and target genes, we predicted peak-gene links through integrated scATAC-seq and scRNA-seq analysis (Fig. 3D). We calculated correlations between peak accessibility and gene expression from the integrated dataset. In parallel, we constructed a co-accessibility network for distal peaks and the promoters of target genes. Confident peak-gene links are contingent on significant peak-gene correlation and a high co-accessibility score (Fig. 3D). In BCC, EC, OC, and RCC, correlations with NDRG1 expression have p-values of 9.3e-05, 1.4e-04, 2.3e-04, and 1.0e-04, respectively, with co-accessibility scores of 0.24, 0.32, 0.62 and 0.88 (Fig. 3E). However, in BC, LC, and PLC, the target region displays a low correlation with the expression of NDRG1 (p-value = 0.25, 0.34, and 0.19). These findings suggest that the target region serves as a potential CRE of NDRG1 in BCC, EC, OC, and RCC, while displaying no regulatory effect on NDRG1 in BC, LC, and PLC. Intriguingly, subsequent examination of DARs across distinct tumor cells indicates a similarity with all exhibiting weak accessibility in the target region for BC, LC, and PLC (Supplementary Fig. S4E–G).
Furthermore, we systematically estimated the proportion of regulatory regions identified by gene regulatory networks relative to DARs of different cell types across diverse cancer types. Notably, five types of cells exhibited more than 500 DARs, of which approximately 10 to 60% were identified as candidate regulatory DNA elements (Supplementary Fig. S4H).
Cell-type-associated epigenetic regulation in different carcinomas
Using the approach described above, we identified 20,109, 11,955, 38,292, 39,640, 18,976, 15,202, 10,140, and 13,368 unique peak-gene link pairs in the single-cell data from BC, BCC, CC, EC, LC, OC, PLC, and RCC, respectively (Fig. 4A). The number of peak-gene link pairs correlates with the number of cells from single-cell ATAC-seq data (Fig. 4B). By examining the overlap between these linked peaks and the DARs identified in each cell type, we identified cell-type-associated regulatory regions (Fig. 4C), which can reveal the specific cell functions of cancer genetic risk loci. For instance, SNP rs4554811 and rs704417 exhibit predicted interactions with nearby transcript promoter regions, indicative of a potential regulatory role for TCF7L1 and PRICKLE2 (Supplementary Fig. S5A, B).
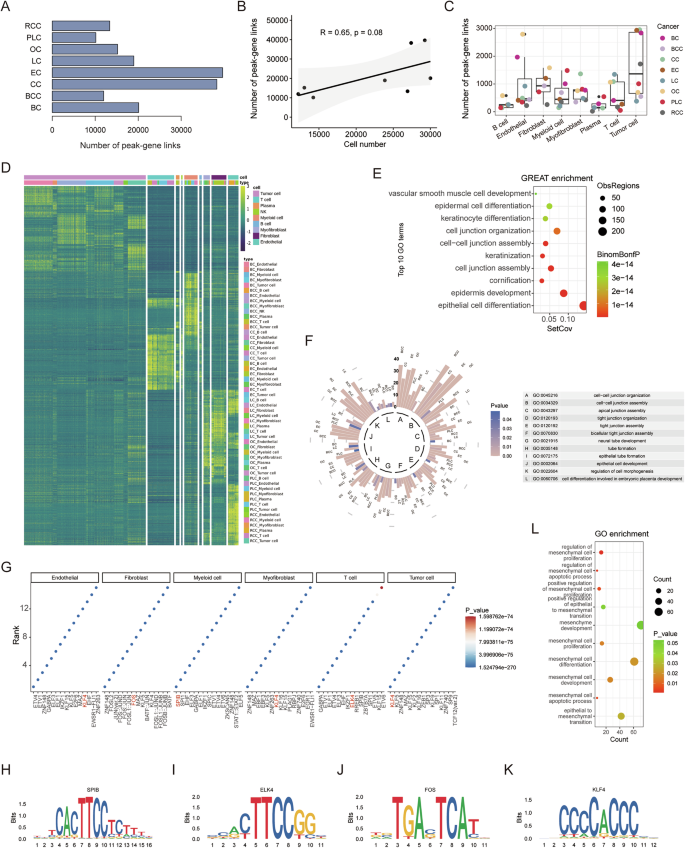
A Number of identified peak-gene links in the eight carcinomas. B The scatter plot and correlation between the cell number in scATAC-seq data and the number of peak-gene links (R represents Pearson’s correlation and its coefficient of determination, the p-value is 0.08). C Number of cell-type-associated peak-gene links in the eight carcinomas. D Heatmap representation of the cell-type-conserved regulatory elements predicted. Each row represents an individual scATAC-seq peak. Color represents the scATAC-seq log normalized accessibility for each peak. E GREAT ontology enrichment analysis of cell-type-conserved regulatory elements. F Circular bar plot of twelve overlapping pathways in the eight carcinomas. The length of the bar represents the number of genes in the indicated pathway, and the color intensity of the bar indicates the p-value. G Enriched motifs in different cell types. Motifs are ranked by p-values from smallest to largest. Motif plots of SPIB H, ELK4 I, FOS J, and KLF4 K. L Gene Ontology enrichment analysis of KLF4 regulatory genes about EMT-related pathways.
We further identified cell-type-conserved regulatory regions in different carcinomas, recognized as candidate regulatory elements not specific to a single carcinoma (Fig. 4D). Tumor cells, T cells, myeloid cells, myofibroblasts, fibroblasts, and endothelial cells present substantial conserved regulatory regions. To assess the potential functions of conserved epigenetic regulation in tumor cells, we employed GREAT ontology enrichment analysis on conserved chromatin regions and gene ontology (GO) enrichment analysis on their target genes. The results revealed that tumor-conserved regions predominantly regulate cell junction and epithelial cell development-related pathways (Fig. 4E, Supplementary Fig. S5C). Moreover, we individually inspected the functions of tumor cell-associated regulatory elements by performing GO enrichment analysis on their predicted regulatory genes. Twelve processes were enriched in all tumor cells, including pathways related to cell junction and epithelial cell development, consistent with previous findings (Fig. 4F, Supplementary Fig. S5D). We also explored the potential functions of conserved regulatory regions in other cell types. For instance, T cells mainly regulate T cell activation, myeloid cells influence the immune response, fibroblasts contribute to extracellular matrix organization, and both myofibroblasts and endothelial cells play roles in blood vessel development (Supplementary Fig. S5E, F).
We next applied motif enrichment analysis on cell-type-conserved regulatory regions to reveal key TFs of each cell type (Fig. 4G). Myeloid cells and T cells are enriched for the binding sites of expected cell-type-activated TFs, such as SPIB and ELK4. Fibroblasts, on the other hand, showed a strong enrichment for AP-1 TF binding sites (Fig. 4H, J). Notably, tumor cells exhibit an enrichment in binding sites for Krüppel-like factors, similar to endothelial cells and myofibroblasts. We identified tumor cell-conserved regulatory regions with KLF4 binding sites and performed GO enrichment analysis on the target genes of KLF4. This analysis revealed that genes regulated by KLF4 are significantly enriched in ontology terms related to epithelial-mesenchymal transition (EMT) (Fig. 4K, L), indicating that KLF4 plays a pivotal role in regulating genes associated with mesenchymal characteristics, consistent with previous studies [56].
Transcription factor activities in different cell types
In prior studies, which elucidated the role of small sets of TFs in governing gene expression programs in tumor cells [57, 58], we sought to identify pivotal TFs participating in oncogenic transcriptional programs. To achieve this, we implemented a filtering strategy to identify highly specific TFs enriched across different cell types in each cancer type. Initially, we calculated the differentially enriched motifs through chromVAR bias-corrected scores for all cell types, selecting those highly enriched across diverse cell types. Subsequently, utilizing footprint analysis, we assessed the binding status of TFs across the genome for different cell types. In each cell type, TFs with a higher degree of activity and binding than those in other cell types were identified as cell-type-associated TFs (Fig. 5A, Supplementary Table S7). Employing UMAP with bias-corrected chromVAR scores for identified cell-type-enriched motifs, we effectively clustered single cells into distinct cell types, illustrating the efficacy of our strategy in screening for cell-type-associated TFs (Fig. 5B).
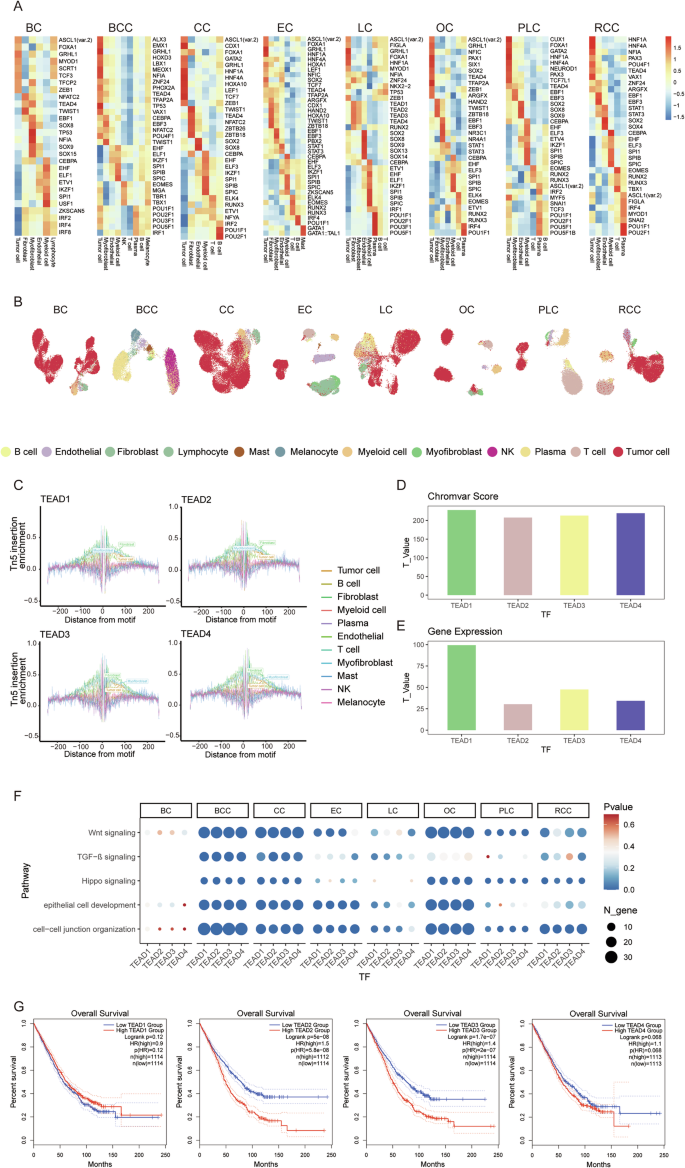
A Heatmap illustrating chromVAR bias-corrected deviation scores for the differential TF motifs of various cell types in eight different carcinomas. B UMAP visualization of single cells based on chromVAR bias-corrected deviations for identified motifs. C TF footprinting of TEAD TFs in diverse cell types in the integrated scATAC-seq data. D Bar plot of TEAD TFs analyzed by comparing the chromVAR score of tumor cells with other cell types. E Bar plot of TEAD TFs analyzed by comparing the gene expression of tumor cells with other cell types. F Dot plot of TEAD TFs regulating cancer-associated pathways in the eight carcinomas. The size of the dot represents the number of TF regulatory genes, and the color intensity of the dot indicates the p-value. G Kaplan-Meier survival analysis of TCGA cancer (COAD, LIHC, LUAD, KIRC, OV, and UCEC) patients with high (top 50%) and low (bottom 50%) expression for the TEAD1/2/3/4 gene.
We made an intriguing discovery that AP-1 TFs exhibit extensive binding across different cell types, with elevated activity in tumor cells, fibroblasts, myofibroblasts, and endothelial cells (Supplementary Fig. S6A, B). To evaluate the impact of AP-1 TFs on gene expression regulation, we mapped their binding sites along with those of 588 other TFs in pairs-linked regulatory regions. Our findings revealed that AP-1 TFs exhibit a more extensive binding pattern compared with other TFs, indicating their broader regulation of gene expression (Supplementary Fig. S6C).
Our investigations also revealed that the identified cell-type-associated TFs in multiple cancer types are similar. For instance, EOMES and TWIST1 are highly activated in T cells and fibroblasts, respectively, indicating a high degree of reliability and consistency. We further performed GO enrichment analysis on the regulatory genes of EOMES in different carcinomas individually, based on the identified peak-gene link networks. The results show that EOMES is predominantly involved in the regulation of T cell activation and other immune-related pathways (Supplementary Fig. S7A). Similarly, TWIST1 is involved in regulating extracellular matrix organization and mesenchyme development, correlating with expected fibroblast functions (Supplementary Fig. S7B). However, distinct TF activity profiles emerged from analyses of different tumor cells. Notably, TEAD1/2/3/4 are activated in tumor cells across most cancer types, except for BC, displaying higher binding status and expression levels compared to other cells in the integrated scATAC-seq and scRNA-seq data (Fig. 5C–E). TEAD family TFs are critical regulators of various oncogenic pathways, including the Hippo signaling pathway, Wnt signaling pathway, and TGF-β signaling pathway. Our investigation revealed that TEADs significantly regulate these pathways in various tumor cells (Fig. 5F). They also participate in the regulation of conserved regulatory pathways in tumor cells, such as cell junction and epithelial cell development. Additionally, overexpression of TEAD1/2/3/4 is significantly linked to poorer overall survival across six cancers (COAD, LUAD, LIHC, KIRC, OV, and UCEC) in TCGA data (Fig. 5G). Across all available TCGA datasets, TEAD family TFs consistently correlate with poorer outcomes, highlighting their consistent impact on different tumor cells and suggesting their potential as prognostic biomarkers (Supplementary Fig. S7C).
Tumor-specific transcription factors regulate malignant transcriptional programs in colon cancer
We found that certain previously identified tumor cell-associated TFs are highly activated in normal epithelial. For instance, HNF4A exhibits high activity in colon and kidney epithelium [13, 59], while TFAP2C has been reported to play a crucial role in promoting epithelial gene expression [60]. To identify tumor-specific TFs, we conducted a comprehensive analysis of scATAC-seq and scRNA-seq data from normal colon tissues (Fig. 6A, B, Supplementary Fig. S8A, B). Five TFs (i.e. CEBPG, LEF1, SOX4, TCF7, and TEAD4) display higher chromVAR scores, increased binding, and elevated expression in tumor cells compared to normal epithelial cells (Fig. 6C, D, Supplementary Table S8).
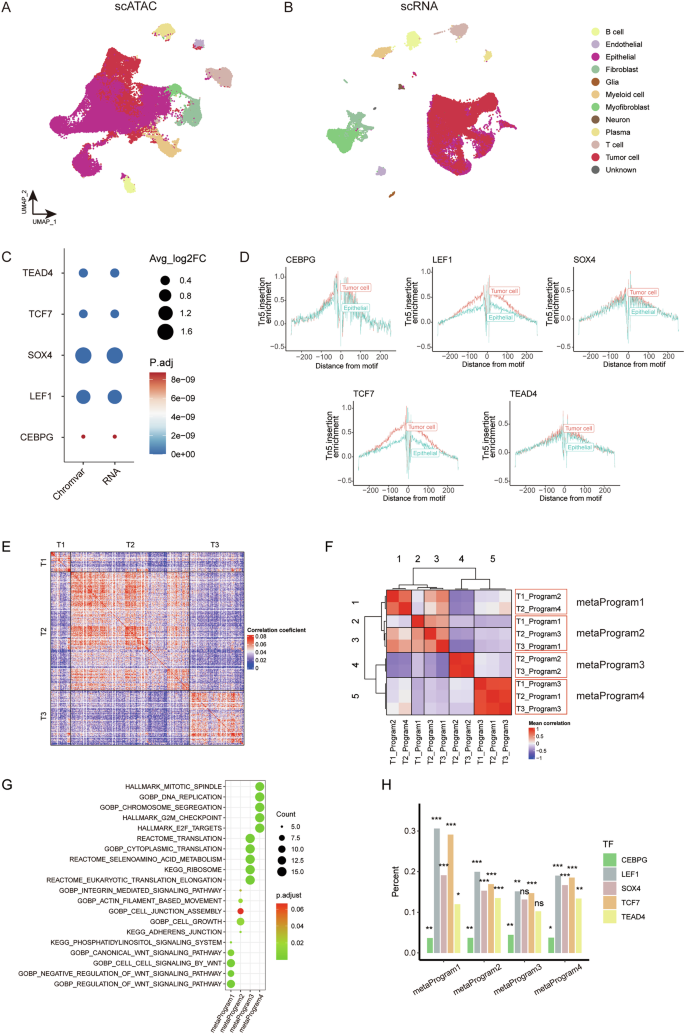
A Integrated UMAP projection based on scATAC-seq data, encompassing all cells from CC tissues and normal colon tissues. B Integrated UMAP projection based on scRNA-seq data, encompassing all cells from CC tissues and normal colon tissues. C Dot plot presenting changes of chromVAR bias-corrected deviation scores and gene expression between tumor cells and normal epithelial cells for the identified colon tumor-specific TF motifs. D TF footprinting of CEBPG, LEF1, SOX4, TCF7, and TEAD4 motifs in CC cells and normal colon epithelial cells. E Pairwise correlations between the expression profiles of three scRNA-seq CC samples. F Heatmap of average correlations across three samples with CC between pairs of programs. The red box indicates the four meta-programs. G Dot plot displaying significantly enriched pathways for signature genes of each meta-program. The dot size is proportional to the relative number of genes in the indicated pathway, and the color intensity indicates the p-value after FDR correction. H Proportion of the five identified tumor-specific TF motifs found in the regulatory elements for the signature genes for each of the four meta-programs (*p p p
To better understand the regulatory landscape driven by the five tumor-specific TFs (i.e. CEBPG, LEF1, SOX4, TCF7, and TEAD4) in CC cells, we utilized scRNA-seq data from CC tissues. We began with a pairwise correlation analysis, which revealed distinct transcriptional states consistently present within these tumors (Fig. 6E). Using nonnegative matrix factorization (NMF), we sought to elucidate the underlying transcriptional programs comprising co-expressed genes, and ultimately extracted 10 malignant transcriptional programs from the scRNA-seq data derived from three patients with CC (Supplementary Fig. S8C). Subsequent hierarchical clustering led to the identification of four meta-programs that encompassed highly similar programs across the three patient samples (Fig. 6F, Supplementary Fig. S8D-E, Table S9). Genes within meta-program 1 exhibit significant enrichment in the WNT signaling pathway, while meta-program 2 is predominantly enriched in pathways related to cell junction. Meta-program 3 and 4 showed enrichment in pathways associated with protein synthesis and DNA replication, respectively (Fig. 6G). To assess the impact of meta-programs on cellular viability in tumor cells, we evaluated the effect of loss-of-function of the top 100 genes in each meta-program on tumor cell viability, by examining CRISPR-Cas9 screening data from The Cancer Dependency Map Consortium (DepMap). Dependency scores (CERES) are normalized, with scores lower than 0 indicating essential genes for the given cell line. Notably, the mean CERES of most genes from 33 CC cell lines are lower than 0, suggesting they are essential in colon tumor cells (Supplementary Fig. S8F).
Next, we reviewed the top 100 genes within these four meta-programs and examined their linked peaks. Notably, motifs of the five tumor-specific TFs were significantly enriched in these peaks, indicating their key role in regulating gene expression in colon tumor cells (Fig. 6H). We further performed gene ontology enrichment analysis on the perturbagen’s signature of CEBPG, LEF1, SOX4, TCF7, and TEAD4, and the results reveal that the five tumor-specific TFs present extensive regulation on meta-program related pathways, consistent with the analysis before (Supplementary Fig. S8G).
We further investigated the regulatory interactions among CEBPG, LEF1, SOX4, TCF7, and TEAD4 in colon cancer cells. Our constructed regulatory network in colon cancer revealed that LEF1 is under the regulation of SOX4 and TCF7, whereas SOX4 is regulated by LEF1, TCF7, and TEAD4. Additionally, CEBPG is regulated by LEF1, TEAD4, and SOX4, while TEAD4 is regulated by LEF1 and TCF7 (Supplementary Fig. S9A). These regulatory dynamics are further confirmed through TF perturbation experiments (Supplementary Fig. S9A, B). Analyzing the COAD RNA-seq data from TCGA, we observed strong correlations among the expressions of LEF1, SOX4, and TCF7 (Supplementary Fig. S9C). These findings provide evidence of their regulatory relationships, indicating the presence of a transcriptional circuitry among tumor-specific TFs.
The importance of tumor-specific TFs is widely supported
To explore the significance of tumor-specific TFs, we performed single-cell multiome sequencing on paired CC tissues and adjacent normal tissues for two patients (“Patient 1” and “Patient 2”) (Fig. 7A, Supplementary Table S10). Additionally, we curated another dataset comprising scATAC-seq and scRNA-seq from the same patient, comparing tumor and normal samples (“Patient 3”). By integrating both transcriptomic and epigenetic data from the same patient samples, we minimized inter-patient variability (Fig. 7B, Supplementary Fig. S10A–D). Our analysis revealed that CEBPG, LEF1, SOX4, TCF7, and TEAD4 exhibited higher expression levels and increased chromVAR scores in tumor cells compared to normal epithelial cells in the three patients (Fig. 7C). We also assessed their binding status across the entire open chromatin genome in tumor and epithelial cells, observing notably increased binding in tumor cells (Fig. 7D, Supplementary Table S8). Moreover, we integrated scATAC-seq data from eight colon tumor samples and seven normal colon samples, along with scRNA-seq data from twenty-three colon tumor samples and ten normal colon samples, which are collectively labeled as “Validation” (Supplementary Fig. S10E–H). The tumor-specific TFs with high activity in colon tumor cells, including CEBPG, LEF1, SOX4, TCF7, and TEAD4, exhibited elevated expression, higher chromVAR scores, and increased binding compared to normal epithelial cells (Supplementary Fig. S10I, J, Table S8). Further analysis using the TCGA and GTEx datasets confirmed that these TFs are expressed at higher levels in tumor tissue samples compared to normal tissue samples (Fig. 7E).
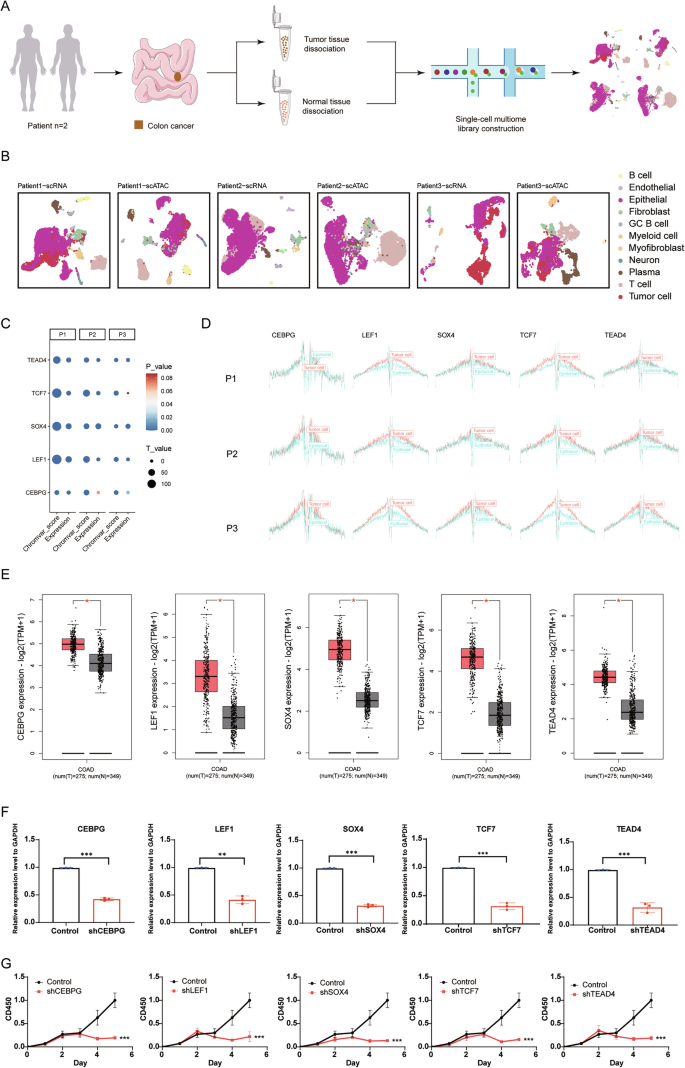
A Schematic representation of the single-cell multiome sequencing experiments in this study. B Integrated UMAP projection based on scRNA-seq and scATAC-seq data, encompassing all cells from CC tissues and normal colon tissues of 3 patients. C Dot plot presenting changes of chromVAR bias-corrected deviation scores and expression levels for the identified colon tumor-specific TFs across tumor cells and normal epithelial cells. D TF footprinting of CEBPG, LEF1, SOX4, TCF7, and TEAD4 motifs in CC cells and normal colon epithelial cells for three patients. E Expression levels of the identified colon tumor-specific TFs in CC tissue samples (orange) and normal tissue samples (gray) obtained from the GEPIA2 database (*p F Downregulation of CEBPG, LEF1, SOX4, TCF7, and TEAD4 mRNA expression in the DLD1 cell line by shRNA knockdown. Statistically significant differences are determined by two-way ANOVA (*p p p G Effects of sh-CEBPG, sh-LEF1, sh-SOX4, sh-TCF7, sh-TEAD4, and vector (control) knockdown on DLD1 cell proliferation as determined by a cell proliferation assay. Statistically significant differences are determined by two-way ANOVA (*p p p
To further our understanding and validate the biological functions of these tumor-specific TFs, we carried out targeted knockdown experiments (Fig. 7F, Supplementary Fig. S11A). The results were striking, showing a substantial reduction in tumor cell proliferation by 5 days after TF knockdown (Fig. 7G). Moreover, migration was reduced, and apoptosis was induced in DLD1 cells with tumor-specific TF deletion, compared to control cells (Supplementary Fig. S11B, C). To identify potential drugs targeting these TFs, we took advantage of the LINCS consortium, which contains 19,811 small-molecule compound-perturbed profiles [47]. Through careful screening, we identified four pharmacological agents—tacedinaline, quinoclamine, dorsomorphin, and vorinostat—each targeting CEBPG, LEF1, SOX4, and TEAD4, respectively. Consistent with our expectations, these four compounds substantially attenuated both the expression levels of CEBPG, LEF1, SOX4, and TEAD4, and affected malignant phenotypes in the DLD1 cell lines, including proliferation, migration, and apoptosis (Supplementary Fig. S11D–H).