To characterize the genetic structure of the Gütersloh outbreak, we attempted to sequence SARS-CoV-2 from 2,241 archived SARS-CoV-2 positive diagnostic swab samples; 1,822 of these were collected from Gütersloh MPP workers for serial diagnostic testing and screening purposes (“outbreak samples”) and 419 collected from Gütersloh-area cases for routine diagnostic testing by Labor Krone (“community samples”). The sample collection dates of the MPP worker samples ranged from 05 June to 23 July 2020, with the large majority (91%) of samples collected during the peak period of the outbreak from mid- to late June. For community samples, sample collection dates ranged from 11 March to 10 October 2020. Of note, the definition of community samples as collected independently of serial screening and testing efforts in the MPP did not preclude the presence of epidemiological connections between the corresponding cases and the MPP, which we therefore investigated separately based on Gütersloh Health Authority data where relevant to our key conclusions (see sections below).
Sequencing was deemed successful if the reconstructed viral genome had ≤ 3000 undefined characters (“Ns”). The final set of analyzed sequences comprised 1,595 viral genomes, of which 1,438 genomes were from outbreak samples. This corresponded to an approximate outbreak case coverage of 68% (calculated as 1,438 / 2,119); the other 157 genomes were from community samples.
Clonality of the Gütersloh MPP outbreak
We compared the sequenced outbreak samples to the reference genome from Wuhan24 (MN908947.3) using Nextclade23. Across all outbreak samples, we detected 529 unique substitutions (mean number of mutations per sample = 8.9 +/− 1.1). Eight substitutions (C241T, C1059T, C2027T, C6406T, C14408T, G18972A, A23403G and G25563T) were present in at least 98.9% and up to 100% (A23403G) of outbreak samples (Fig. 1B). Furthermore, all eight substitutions were present in 98.2% (n = 1412) of outbreak samples. Nearly 42% of outbreak samples (n = 602) contained these eight and no other substitutions. We thus declared these substitutions as the “outbreak-defining substitutions”. Furthermore, 1,424 outbreak samples (99.0%) were assigned to lineage B.1.329, while the remaining 14 (1.0%) were assigned to three other lineages; we therefore declared B.1.329 as the outbreak lineage (Fig. 1A). The analysis of individual mutation patterns and the lineage-level analysis thus indicated that the outbreak was clonal. Moreover, the frequency of cases potentially associated with other viral lineages remained below 2%, indicating an at most minor contribution of secondary introduction events to the overall case load of the outbreak.
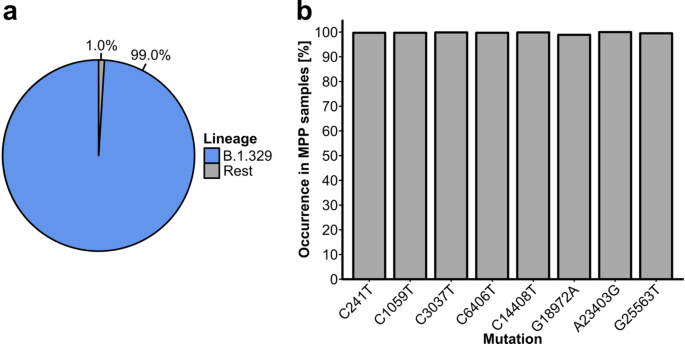
Presence of B.1.329 and outbreak-defining substitutions in sequenced outbreak samples. (a) Lineage assignment of the sequenced outbreak samples. (b) Frequency of the outbreak-defining substitutions in the sequenced outbreak samples.
Presence of B.1.329 in sequenced community samples
We investigated the frequency of the outbreak-associated lineage B.1.329 in sequenced community samples, i.e. in samples collected from Gütersloh-area cases for routine diagnostic testing purposes (Fig. 2). B.1.329 was first detected in a community sample collected in calendar week 12 of 2020, i.e. 14 weeks prior to the peak of the MPP outbreak in calendar week 26. The B.1.329 strain circulated in the community until at least calendar week 18, followed by a three-week period (weeks 19–21) without any sequenced community samples. Between calendar weeks 22 and 28, corresponding to the period a few days after the occurrence of superspreading in the Gütersloh MPP to approximately two weeks after the peak of the outbreak (25 May–12 July), B.1.329 accounted for 60% to 100% of the sequenced community samples. Afterwards, B.1.329 was not detected in the community for an 8-week period, until it briefly re-appeared at low absolute sample counts over a 3-week period in the fall.
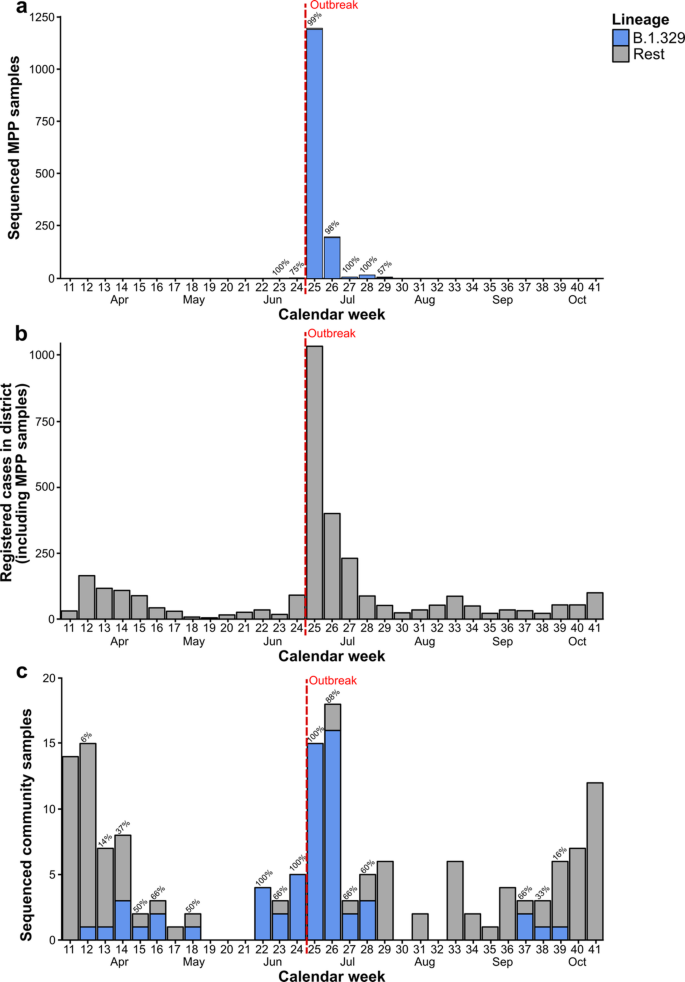
Weekly case numbers in the MPP and the surrounding district. (A) Sequenced outbreak samples stratified by lineage and calendar week. (B) Registered cases in Gütersloh district by calendar week (including outbreak cases). (C) Sequenced community samples by lineage and calendar week. Percentages above the bars indicate the proportion of sequenced cases assigned to the outbreak lineage B.1.329, if applicable. Community samples were defined as samples collected from Gütersloh-area cases for routine diagnostic testing purposes.
The detection of B.1.329 in terms of the absolute numbers of B.1.329-carrying sequenced community cases generally mirrored the short-term outbreak-associated increase in overall SARS-CoV-2 cases in Gütersloh between calendar weeks 24 and 28, while also showing community circulation in the periods before and after the outbreak. Outside of the outbreak period, SARS-CoV-2 case numbers in Gütersloh generally followed the development of case numbers in Germany over the course of 202025, exhibiting a spike in case numbers in the spring, a decrease during the summer, and another period of increase in the fall (Fig. 2A). Similarly, the clade distribution of sequenced community samples collected outside of the outbreak period was similar to that observed across Germany (Supplementary Fig. 2). In terms of Pango lineages, we detected a total of 9 different lineages (including the outbreak lineage B.1.329), most of which were sub-lineages of B.1, in the pre-outbreak period; in the post-outbreak period, we detected a total of 21 lineages, including the outbreak lineage (Supplementary Table 1).
Prior circulation of the outbreak-associated strain in the community, independent of the MPP
To investigate the potential community circulation of the outbreak-associated strain in the pre-outbreak period, we first carried out a fine-scale mutational analysis of the 20 B.1.329-carrying community samples collected prior to the peak of the outbreak in June 2020 (Fig. 3). Nine B.1.329-carrying samples were collected before May 2020 (calendar week 18); of these, three carried all eight outbreak-defining substitutions and no others. Furthermore, based on case and contact tracing data collected by Gütersloh Health Authority, we investigated 15 of the 20 identified B.1.329 community samples for potential epidemiological connections between the cases that these samples were collected from and the MPP (Fig. 3); of note, this set included all identified B.1.329-carrying samples with collection dates before May 2020. For 11 of the 15 samples investigated, we identified an epidemiological connection between the corresponding cases and the MPP; the earliest sample collection date in this set was 26 May. For the remaining four samples, no epidemiological connections between the corresponding cases and the MPP were detected; two of these, collected in early April (Fig. 3; sample IDs 1,055,342,199 and 1,055,342,331), carried exactly the set of eight outbreak-defining mutations. In conclusion, we detected the outbreak-associated strain in community samples without any apparent connections to the MPP more than six weeks prior to the initial transmission of the outbreak strain to Gütersloh MPP workers.
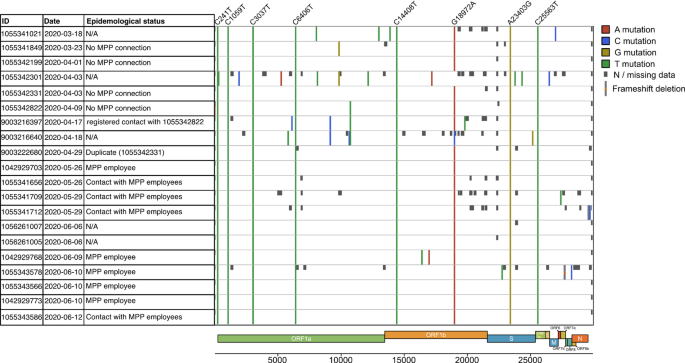
B.1.329 community samples collected prior to the outbreak. The first three columns show sample ID, sample collection date, and whether case and contact tracing data indicated any epidemiological links between the sample and the MPP. The samples were compared to the Wuhan reference genome using Nextclade. Colored lines indicate SNPs compared to the reference genome, the color indicating the detected variant allele differing from the reference. The eight outbreak-defining mutations are listed on the top. Missing data is represented by dark gray boxes. Light gray lines with a red square indicate a frameshift deletion. Parts of this figure are based on the result output from Nextclade.org23.
No indication of the presence of B.1.329 in other countries
We carried out a GISAID26 search for B.1.329 and identified 1,686 samples (Supplementary Table 2); 1,626 of these were submitted by us as part of this project. All identified samples were collected in 2020 and none of the identified samples were collected in countries other than Germany (Supplementary Fig. 3; see Supplementary Table 2 for details). Of the 60 B.1.329 samples found in GISAID that were not part of this study, 35 were uploaded by Günther et al.; the remaining 25 samples were uploaded by the University of Bielefeld, had sample collection dates between 08 June 2020 to 20 June 2020, and were collected in the city of Bielefeld, also located in the German state of North-Rhine Westphalia, approximately 20 km from Gütersloh. Based on this information, the earliest detection of B.1.329 was in the community samples sequenced as part of this study.
Limited evidence for the persistence of multiple viral sub-lineages in the peak period of the outbreak
To determine the extent to which the fifteen viral sub-lineages (“haplotypes”; see Methods and Supplementary Fig. 1) present among the 35 SARS-CoV-2 genomes sequenced by Günther et al.10 persisted into the peak period of the outbreak, we searched our set of MPP samples for exact matches to these. For five sub-lineages, named after the ID of the first samples carrying the corresponding haplotype in Günther et al., at least one exact match was detected. For sub-lineage B1, defined by the set of 8 outbreak-defining mutations, 602 exact matches in our MPP dataset were detected; the corresponding sample B1 was collected from one of the two MPP workers involved in the initial transmission of the outbreak strain into the population of Gütersloh MPP workers. For sub-lineages O9, O7, and O2, we detected 23, four, and three matches, respectively; the corresponding samples were collected in mid-June and thus fell into the period during which the majority of our MPP samples were collected. Finally, we detected one exact match to sub-lineage B2; the corresponding sample was collected from the other MPP worker involved in the initial transmission of the outbreak strain into the population of Gütersloh MPP workers. In conclusion, of the sub-lineages present in samples collected during the early outbreak period in May 2020 in the Günther et al. analysis, apart from a single match to B2, only B1 was found to have persisted into the peak period of the outbreak.