Experiments performed in this study did not require ethics board approval.
Cell culture
HAP1 cells were cultured in Iscove’s modified Dulbecco’s medium (IMDM) supplemented with GlutaMAX (Thermo Fisher Scientific), 25 mM HEPES, 10% FBS and 1% penicillin–streptomycin, following standard procedures. Cells were routinely checked and sorted for haploidy. All 293TX cells were cultured in DMEM supplemented with 10% FBS and 1% penicillin–streptomycin.
Antibodies
Antobidoes used included Anti-SMC1 (A300-055A, Bethyl), anti-SMC3 (A300-060A, Bethyl), anti-RAD21 (05-908, Merck), anti-NIPBL (A301-779A, Bethyl), anti-FLAG (F1804, Merck), anti-SCC4/MAU2 (ab183033, Abcam), anti-GAPDH (sc-32233, Santa Cruz), anti-STAG1 (A302-579A, Bethyl), anti-STAG2 (A300-159A, Bethyl), anti-CTCF (ab128873, Abcam), anti-H3K4me3 (39060, Active motif), anti-H3K27ac (39133, Active motif), anti-V5 (R960-25, Thermo Fisher Scientific), anti-WAPL (sc-365189, Santa Cruz) and anti-PDS5A (A300-089A, Bethyl).
Plasmid construction
The plasmids expressing TetR–FLAG–MAU2 and TetR–FLAG–mCherry cassettes were cloned into a lentivirus backbone under the control of the EF1 promoter. TetR, FLAG and MAU2 or mCherry sequences were PCR-amplified with 20 bp overhang for In-Fusion cloning. The final expression cassette comprised EF1-TetR-FLAG-MAU2/mCherry-P2A-Puromycin. To construct the V5–MAU2 plasmid, the TetR–FLAG sequence from the TetR–FLAG–MAU2 construct was removed, and a V5 tag was inserted instead. To enable simultaneous expression of the two MAU2 constructs, the antibiotic selection marker was replaced by blasticidin instead of puromycin. To insert the AID2 tag into the endogenous gene, a single guide RNA (sgRNA) targeting the ORF of the gene was cloned into a vector containing SpCas9–T2A–BFP (Supplementary Table 1). To construct the donor template for AID2 tag insertion, a cassette containing AID2–GFP was cloned between two homology arms of about 1 kb surrounding the sgRNA cut site. Detailed plasmid maps can be found in Supplementary Information.
Generation of cell lines containing the TetO platforms
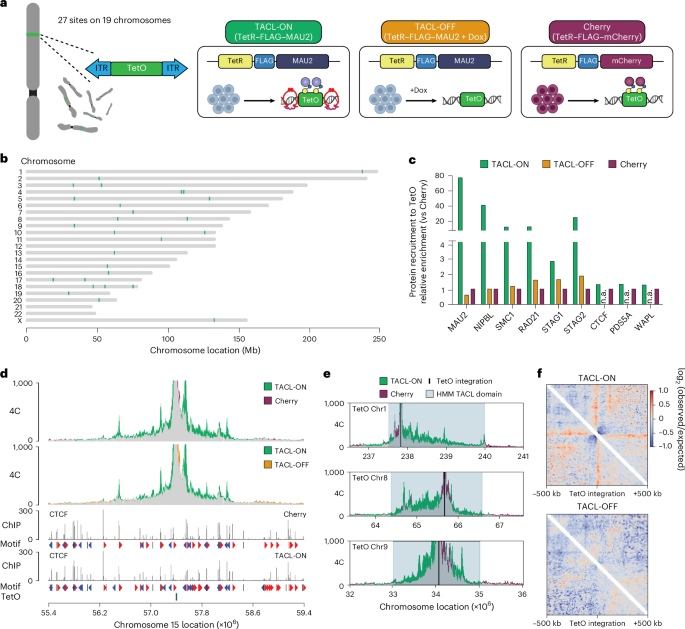
The plasmids bearing the TetO platforms and the PiggyBac transposase were originally obtained from L. Giorgetti39, validated by Nanopore sequencing with 48× repeats (see Supplementary Table 2 for sequences). In brief, HAP1 cells were trypsinized and resuspended in serum-free IMDM medium. A vector containing the PiggyBac transposase (pBroad3_hyPBase_IRES_tagRFPt) was mixed with a PiggyBac donor vector bearing 30× TetO binding sites and polyethylenimine (PEI; Polysciences) in serum-free IMDM. The DNA mix was incubated at room temperature (20–22 °C) for 10 min, after which the cells and the DNA mix were incubated together for another 10 min. The cells were then plated in a six-well plate. After 24 h, the medium was refreshed. Then, 48–72 h after the transfection, the cells were sorted for a RFP signal, expressing the transposase. Sorted cells were plated in a 15 cm dish and cultured for at least 14 days. Colonies were picked and sub-cultured in 96-well plates. To genotype the clones with a sufficient number of integration sites, cells were lysed in DirectPCR lysis reagent (Viagen). Lystes were subsequently assessed by running qPCR with primers annealing to the transposon sequences. A primer targeting a part of the human FSIP2 gene was used as the reference among different clones. An estimation of the number of integration sites was calculated as: \({2}^{-({\mathrm{Ct}}_{{\rm{T}}{\rm{e}}{\rm{t}}{\rm{O}}\,{\rm{p}}{\rm{r}}{\rm{i}}{\rm{m}}{\rm{e}}{\rm{r}}}-{\mathrm{Ct}}_{\mathrm{Re}{\rm{f}}{\rm{e}}{\rm{r}}{\rm{e}}{\rm{n}}{\rm{c}}{\rm{e}}})}\). The exact number of integration sites was validated by 4C-seq.
Lentivirus production and transduction
A total of 4 × 106 293TX cells were plated in a 10 cm dish 24 h before virus production. Lentiviral vectors were co-transfected with pVSV-G, pMDL RRE and pRSV-REV in serum-free DMEM with PEI (Polysciences). The medium was refreshed 18 h after transfection. The medium containing the virus particles was collected 48 h after transfection by passing through a 0.45 μm filter. For transduction, HAP1 cells were plated in a six-well plate 24 h before transduction. The transduction was performed by adding the virus particles directly onto the cells supplemented with 6 μg ml−1 polybrene (Merck). The cells were refreshed 24 h after transduction, and antibiotics (puromycin and blasticidin) were added 48 h after transduction. Cells were selected with antibiotics until the cells in the control plate (without transduction) were completely dead.
Western blot
Cells were washed in PBS and lysed in RIPA buffer with protease inhibitor (Roche) on ice for 15 min. The cell lysate was further disrupted by sonication with Bioruptor Pico (Diagnode). The cell lysate was cleared by spinning at 1,000g for 5 min. The supernatant was incubated with Laemmli buffer and boiled for 10 min. The sample was then loaded on a 4–15% Mini-PROTEAN TGX Precast Protein Gel (Bio-Rad) and run at 100 V for 90 min. Proteins were transferred onto a nitrocellulose or PVDF membrane and incubated with the primary antibody overnight at 4 °C. The membrane was then washed in PBS with 0.25% Tween and incubated with the secondary antibody at room temperature for 1 h. Finally, the membrane was incubated with SuperSignal West Pico PLUS Chemiluminescent Substrate (Thermo Fisher Scientific) for 1 min before being visualized on ImageQuant 800 imager (Amersham).
Nuclear and cytoplasmic fractionation
In brief, 3 × 106 cells were collected by trypsinization. Cells were washed with PBS, and the cell pellet was resuspended in 100 μl of cytoplasmic extraction buffer (10 mM HEPES, 60 mM KCl, 1 mM EDTA, 0.075% (v/v) NP-40, 1 mM dithiothreitol and 1 mM PMSF, final pH 7.6) and incubated on ice for 3 min. The suspension was spun at 1,500 rpm for 4 min, and the supernatant was kept as the cytoplasmic fraction. The pellet was washed once with cytoplasmic extraction buffer. The cells were pelleted at 1,500 rpm for 4 min and resuspended in 50 μl of nuclear extraction buffer (20 mM Tris Cl, 420 mM NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 1 mM PMSF and 25% (v/v) glycerol, final pH 8.0). The salt concentration was adjusted to 400 mM NaCl, and an additional pellet volume of nuclear extraction buffer was added. The pellet was vortexed and incubated on ice for 10 min. The suspension was spun at max speed for 10 min, and the supernatant was kept as the nuclear fraction.
ChIP
A total of 100 million cells were crosslinked with 1% formaldehyde for 10 min. Cells were subsequently quenched with 125 mM glycine for 10 min and washed twice with cold PBS. Cells were scraped from culture dishes, and cell pellets were subsequently lysed in LB1 buffer (50 mM HEPES, 140 mM NaCl, 1 mM EDTA, 10% glycerol, 0.5% NP-40, 0.25% Triton X-100), washed in LB2 buffer (10 mM Tris, 200 mM NaCl, 1 mM EDTA, 0.5 mM EGTA) and resuspended in LB3 buffer (10 mM Tris, 100 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 0.1% sodium deoxycholate, 0.5% N-lauroylsarcosine) before sonication. Chromatin was sonicated using Bioruptor Pico (Diagnode) with a setting of 30 s on, 30 s off for eight cycles. Fragmented chromatin was then incubated with 6 µg of antibodies pre-coupled to Dynabeads Protein G beads (Thermo Fisher Scientific) overnight at 4 °C. Bead-bound chromatin was then washed 10× with RIPA buffer (50 mM HEPES, 500 mM LiCl, 1 mM EDTA, 1% NP-40, 0.7% sodium deoxycholate), once with TBS buffer and decrosslinked in elution buffer (50 mM Tris, 10 mM EDTA, 1% SDS) at 65 °C for 18 h. Eluted DNA was then treated with protease K and RNAse A, and subsequently purified with phenol/chloroform/isoamyl alcohol 25:24:1. Purified DNA was either assessed with qPCR (see Supplementary Table 1 for oligonucleotides used) or continued with ChIP–seq next-generation sequencing library preparation. Sequencing libraries were constructed using the NEBnext Ultra II DNA library prep kit (New England Biolabs, NEB) following the manufacturer’s protocol. In brief, DNA was end-repaired and poly-A tailed, ligated to NEBnext adaptors and digested with USER enzyme. Annealed libraries were then purified with AMPure XP beads (Beckman Coulter) and PCR-amplified with indexing primers for 4–12 cycles. Sequencing libraries were checked with Bioanalyzer HS DNA chip (Agilent) and sequenced on the Illumina NextSeq 500 (single-end reads, 75 bp) and NextSeq 2000 platforms (paired-end reads, 50 bp).
4C-seq
The 4C template preparation was performed as previously described57,58. In brief, ten million cells per sample were crosslinked with 2% formaldehyde, followed by quenching by glycine at a final concentration of 0.125 M. The four-cutter restriction enzyme MboI (NEB) was used for in situ digestion (300 U per ten million cells). Digested DNA fragments were ligated, reverse-crosslinked and subsequently purified through isopropanol and magnetic beads (Macherey–Nagel NucleoMag PCR Beads). The four-cutter restriction enzyme Csp6I (CviQI, Thermo Fisher Scientific, ER0211; 50 U per sample) was used for template trimming. Re-ligated and purified 4C templates were further processed through in vitro Cas9 digestion as described below.
In vitro Cas9 digestion of 4C templates
To prevent PCR amplification and sequencing of TetO repeats owing to tandem ligation of two or more TetO DpnII fragments in a given 4C circle, an in vitro digestion of 4C templates was performed as previously described59 with the following modifications: two sgRNAs were used to target Cas9 into the TetO repeats between viewpoint primers; and pre-incubation of the Cas9 protein and sgRNA template was performed at room temperature. In brief, two sgRNA templates were obtained using the Megashortscript T7 transcription kit (Invitrogen), followed by 4× AMPure XP (Agencourt) purification. Purified Cas9 protein (generated by Hubrecht protein facility) was pre-incubated with the sgRNAs for 30 min at room temperature. The 4C templates were subsequently added to the pre-incubated Cas9–sgRNA complexed for overnight digestion at 37 °C. Cas9 protein was inactivated by incubating at 70 °C for 5 min. The resulting products were purified with 1× AMPure XP and used as a PCR template for TetO-dedicated 4C.
Nascent RNA sequencing (BrU-seq)
BrU-seq was performed as previously described60. Cultured cells were incubated with 2 mM bromouridine (BrU, Merck) for 10 min and subsequently lysed in TRIzol reagent (Thermo Fisher Scientific). RNA was isolated following the manufacturer’s protocol. In brief, lysed cells were mixed with chloroform and centrifuged for 15 min. The aqueous phase was transferred to a new tube and mixed with isopropanol. After centrifugation, the RNA pellet was washed once with 70% ethanol and dissolved in DEPC water. To capture BrU-labeled nascent RNA, 6 µg anti-BrdU antibodies (BD Biosciences) pre-coupled with Dynabeads Protein G beads (Thermo Fisher Scientific) were incubated with the total RNA for 1 h at room temperature. The beads were then washed three times with PBS/0.1% Tween-20/RNaseOUT. To purify the bead-bound RNA, TRIzol reagent was directly added to the beads, and RNA was purified as described above. Next-generation sequencing libraries were generated using the NEBnext Ultra II directional RNA library prep kit (NEB) following the manufacturer’s protocol. In brief, RNA was fragmented to about 200 bp in size. First-strand and second-strand cDNA were synthesized. Double-strand cDNA was then end-repaired, poly-A-tailed, ligated to NEBnext adaptors and digested with USER enzyme. Annealed libraries were then purified with AMPure XP beads (Beckman Coulter) and PCR-amplified with indexing primers for seven cycles. Sequencing libraries were checked with Bioanalyzer HS DNA chip (Agilent) and sequenced on the Illumina NextSeq 2000 platforms (single-end reads, 50 bp).
Hi-C
Hi-C template preparation was performed as previously described25. In brief, ten million cells per sample were crosslinked with 2% formaldehyde, followed by quenching by glycine at a final concentration of 0.2 M. The four-cutter restriction enzyme DpnII (NEB) was used for in situ digestion (400 U per ten million cells). Digested DNA was repaired with biotin-14–dATP (Life Technologies) in a Klenow end-filling reaction. End-repaired, ligated and reverse-crosslinked DNA was subsequently purified using isopropanol and magnetic beads (Macherey–Nagel NucleoMag PCR Beads). Purified DNA was sheared to 300–500 bp with Covaris and subsequently size-selected by AMPure XP (Agencourt). Appropriately sized ligation fragments marked by biotin were pulled down with MyOne Streptavidin C1 DynaBeads (Invitrogen) and prepped for Illumina sequencing.
ATAC-seq
ATAC-seq was conducted following the Omni-ATAC protocol. In summary, 200,000 cells were lysed using a solution containing 0.1% NP-40, 0.1% Tween-20 and 0.01% digitonin, then incubated with a homemade Tagment DNA Enzyme for 30 min at 37 °C. DNA purification was carried out using the QIAGEN MinElute Reaction Cleanup Kit. Library fragments were amplified with Phusion High-Fidelity PCR Master Mix with HF Buffer (Thermo Fisher Scientific, cat. no. F531S) and custom primers featuring unique single or dual indexes. Purification of the libraries was performed using AMPure XP beads (Beckman Coulter, cat. no. A63881), following the manufacturer’s guidelines. The quality of the constructed libraries was assessed using the Agilent Bioanalyzer 2100 with the DNA 7500 kit (cat. no. 5067-1504).
Generation of auxin-inducible degron cells
To deplete the cohesin factors in cells, we used the AID2 system49. For RAD21 degron, we generated HAP1 cells stably expressing OsTIR1 (F74G) by transducing the cells with lentivirus containing an expression cassette of OSTIR1-P2A-hygromycin. After antibiotic selection with hygromycin, cells were co-transfected with a vector expressing an sgRNA against RAD21 and SpCas9–T2A–BFP, and the donor template containing AID-GFP flanked by homology arms. GFP+ cells were analyzed and sorted with flow cytometry. Single-cell clones were expanded and used for downstream analysis. For WAPL, PDS5A, STAG2 and CTCF degrons, we first inserted the AID–GFP cassette by co-transfecting the cells with an sgRNA against each gene. Single-cell clones were selected and verified by PCR. Verified clones were then transduced with lentivirus containing an expression cassette of OSTIR1-P2A-blasticidin. To deplete the proteins, we treated the cells with 1 μM auxin (IAA; BioAcademia) for 2–3 h and analyzed the successful depletion with western blot.
Data analysis4C-seq
4C-seq reads were mapped to the hg38 reference genome and processed using pipe4C57 (https://github.com/deLaatLab/pipe4C). Normalized 4C coverage was calculated separately for each TetO integration site using R (www.r-project.org). Counts at non-blind fragments within a 20 Mb region (10 Mb upstream and downstream of the viewpoint) were adjusted to one million mapped reads after exclusion of the two highest-count fragments. Count data was smoothed using a running mean with a window size of 21 fragments using the R package caTools (v.1.18.2).
Aggregate 4C analysis
In 3C-based assays, ligation frequencies are typically highest near the viewpoint (
TACL domains annotation
To systematically annotate the TACL domains induced by the recruitment of cohesin to the TetO platforms, we developed an HMM. The HMM was implemented using the Python package hmmlearn (https://github.com/hmmlearn/hmmlearn). We created an HMM with the states ‘TACL_domain’ and ‘no_change’. The normalized 4C-seq signals for TACL-ON and Cherry conditions were binarized into two observations: ‘TACL_domain’ (4C-seq signal difference between TACL-ON and Cherry >25) and ‘no_change’ (4C-seq signal difference ≤25). The emission probabilities were estimated using manually defined TACL domains. The probability of the TACL_domain state was calculated as the fraction of restriction fragments with 4C-seq signal >25 in the manually defined TACL domains and set to 0.6. The probability of the no_change state was calculated as the fraction of restriction fragments with 4C-seq signal ≤25 in the flanking regions of the manually defined TACL domains and was set to 0.98. The transition probability was set to 10−6.
The estimated TACL_domain and no_change states were then subjected to several additional filters. First, restriction fragments belonging to stretches of more than 20 consecutive TACL_domain states were retained. Second, restriction fragments with consecutive TACL_domain states within 100 kb of each other were merged. Third, merged regions containing at least 40 restriction fragments were retained and further merged within 1.5 Mb of each other to draft TACL domains. Finally, if TetO was outside of the drafted TACL domain, the closest domain segment on the other side of the domain with respect to the TetO location was added to obtain TACL domains.
HMM model with the same parameters was used to annotate TACL domains in the CTCF–AID, WAPL–AID, STAG2–AID and PDS5A–AID lines by comparing the difference between IAA and Dox treatments. Additionally, the same HMM model was used to annotate STAG2 collapsed domains by comparing the difference between the IAA treatment and the untreated condition in the STAG2–AID line. For the filtering steps, the distance for considering restriction fragments with consecutive TACL_domain states was set to 200 kb, and the distance for drafting TACL domains from restriction fragments was set to 2.5 Mb.
ChIP–seq
HAP1 H3K4me1 data are publicly available (ENCODE: ENCSR450JTP).
ChIP–seq reads were mapped to the hg38 reference genome and processed using the 4DN ChIP–seq pipeline (https://github.com/4dn-dcic/chip-seq-pipeline2). P value signal bigwigs were used for all heatmaps and example plots. For wild-type, T-MAU2, T-MAU2 treated with Dox or T-mCherry cells, the P value signals were normalized based on the average P value signal for all CTCF peaks in TACL-ON (for CTCF, RAD21, SMC1, SMC3, STAG1, STAG2, WAPL, PDS5A), FLAG peaks in TACL-ON (for FLAG, MAU2, NIPBL, V5), H3K27ac peaks (H3K27ac) or H3K4me3 peaks (H3K4me3) located outside the TACL domains and further than 3 Mb from the TetO integration sites. In brief, ChIP–seq peaks were filtered for a ‘signalValue’ that represented clear peaks by visual inspection (CTCF, 35; FLAG–MAU2, 35) and for overlapping peaks, such that for overlapping peaks the peak with the highest signalValue was kept. Filtered peaks were resized to 10 bp, and the signal was calculated using the GenomicRanges and rtracklayer package in R/Bioconductor61,62. The average signal was used as the scaling factor. For degron lines, P value signals were normalized based on the average signal of the regions flanking the filtered peaks. In brief, peaks were filtered for signalValue and for overlapping peaks as described above. Next, peaks were resized to 5 kb, and the signals of the outer 1 kb regions (2.5–1.5 kb upstream and downstream of the peak center) were calculated. The average signal of the outer 1 kb regions was used as the scaling factor. For heatmaps, the signal coverage was calculated per 10 bp bin as described above and normalized using the previously determined scaling factor. For the average ChIP signal plot, the average signal for each 10 bp bin was calculated. TetO enrichment ChIP–seq reads were mapped to the hg38 human reference genome assembly with added minimal PiggyBac TetO sequence (Supplementary Table 2) using bowtie2 (v.2.5.2)63. Alignments with a mapping quality (MAPQ) score of ≥1, either to the PiggyBac TetO sequence or elsewhere in the genome, were quantified using FeatureCounts (v.2.0.6)64. Enrichment levels were determined by comparing the coverage to the average coverage from all input control experiments.
Differential FLAG peaks
FLAG ChIP–seq reads were aligned as single-end reads to the hg38 human reference genome assembly with added TetO sequence using bowtie2 (v.2.5.2)63. Reads with MAPQ ≥ 15 were selected using SAMtools (v.1.15)65, and duplicate reads were removed with the Picard (v.2.25.6) ‘MarkDuplicates’ function (https://broadinstitute.github.io/picard). Coverage over FLAG peaks was then quantified using FeatureCounts (v.2.0.6)64 and normalized with DESeq2 (v.1.38.3)66. For TACL‑ON samples, an average signal was calculated by taking the mean of the two replicates. With the addition of a pseudocount of 1, the log2(fold change) between TACL‑ON and TACL‑OFF conditions was computed. Differential FLAG peaks were defined as those with a log2(fold change) value of >1 and with an average TACL‑ON signal exceeding 24.5.
Classification of CTCF sites
Genome-wide CTCF sites were defined as those CTCF peaks located outside of TACL domains and at least 3 Mb away from any TetO integration site in TACL-ON cells. To stratify these sites by CTCF binding strength, we used the ChIP–seq coverage values in TACL-ON. Peaks with a signal below the 33rd quantile were classified as low, those between the 33rd and 66th quantiles as medium and those above the 66th quantile as high.
The presence and orientation of CTCF motifs below each CTCF peak were identified using FIMO (v.5.3.0)67 using the MA0139.1 motif68 and the parameter –max-stored-scores 50,000,000. CTCF peaks for which all identified motifs were located on the plus strand were classified as forward CTCF peaks, while peaks for which all identified motifs were located on the minus strand were classified as reverse CTCF peaks. Forward CTCF motifs located upstream of TetO sites and reverse CTCF motifs located downstream of CTCF were classified as convergent CTCF binding sites. Reverse CTCF motifs located upstream of TetO sites and forward CTCF motifs located downstream of CTCF were classified as divergent CTCF binding sites.
Analysis of ATAC-seq and ChIP–seq for histone modifications
Data processing
ATAC-seq reads were mapped to the hg38 human reference genome assembly using bwa mem (v.0.7.17-r1188)69. ChIP–seq reads were mapped to the hg38 human reference genome assembly with added TetO sequence using bowtie2 (v.2.5.2)63. Uniquely mapped reads in proper read pairs (-f 2) with MAPQ > 10 and MAPQ ≥ 15 were selected using SAMtools (v.1.15)65 for ATAC-seq and ChIP–seq data, respectively. Duplicate reads were filtered out using the Picard (v.2.25.6) and (v.3.1.1) ‘MarkDuplicates’ function (https://broadinstitute.github.io/picard) for ATAC-seq and ChIP–seq data, respectively. Bigwig coverage tracks were generated using the ‘bamCoverage’ function from the deepTools (v.3.4.2)70 with the ‘–effectiveGenomeSize’ parameter set to 2,913,022,398 and ‘–normalizeUsing’ parameter set to RPGC.
Peak calling
Peaks were called using MACS2 (v.2.2.6)71 for pooled data and replicates in a narrowPeak mode, with mappable genome size set to hs, a q value cutoff of 0.05, ‘–keep-dup’ parameter set to all and the ‘–nomodel’ parameter. The consensus peak list was obtained by overlapping the peaks called for pooled data with peaks from replicates. Only the peaks from canonical chromosomes outside of the blacklist regions72 that had an overlap of at least 50% with peaks from both replicates were retained.
Peak analysis
ATAC-seq and H3K27ac peaks from the TACL-ON, TACL-OFF and Cherry conditions were pooled into one set for differential occupancy analysis. Peak counts were obtained using the ‘intersect’ function from BEDTools (v.2.27.1)73 with ‘-c -wa’ parameters. Differential ATAC-seq and H3K27ac peaks were identified using the DESeq2 (v.1.30.1)66. The ‘nbinomWaldTest’ function with default parameters was used to test contrasts. Peaks with a false discovery rate of 2(fold change) of >0.5 were considered significant. For downstream analyses, peak overlap was performed using Bioframe (v.0.3.0). H3K27ac peaks that overlapped with H3K4me3 peaks were classified as promoter peaks, and non-overlapping peaks were classified as enhancer peaks.
Bru-seq
Data processing
BrU-seq reads were mapped to the hg38 human reference genome assembly using STAR (v.2.7.9a)74 with GENCODE (v.44) gene annotation. Uniquely mapped reads with MAPQ > 10 were selected and split by strand using SAMtools (v.1.12)65. Forward strand reads were extracted by using -f 16 FLAG, and reverse strand reads were extracted by using -F 16 FLAG. Gene counts were obtained using the ‘htseq-count’ function from HTSeq (v.0.13.5)75. Counts were calculated separately for genes from forward and reverse strands with the parameters ‘–stranded no’, ‘–nonunique all’, ‘–order pos’ and ‘–type gene’.
Differential expression analysis
Differentially expressed genes were identified using the DESeq2 (v.1.30.1)66 (Supplementary Table 3). Low-expressed genes were filtered by requiring the samples to have gene counts greater than ten. The ‘nbinomWaldTest’ function with default parameters was used to test contrasts. Genes with a false discovery rate of 2(fold change) of >1 were considered significant. For downstream analyses, the genes were overlapped with annotated TACL domains and split into groups depending on their relative distance and position to the TetO platforms using bioframe (v.0.3.0).
Hi-C analysis
Data processing
Hi-C data was processed using the distiller pipeline from Open2C (https://github.com/open2c/distiller-nf). The reads were mapped to the human reference genome assembly hg38 with bwa mem (v.0.7.17-r1188)69 with ‘-SP’ FLAGs. The alignments were parsed and filtered for duplicates using the pairtools (v.0.3.0)76. The complex walks in long reads were masked with ‘–walks-policy’ set to mask, the maximal allowed mismatch for reads to be considered as duplicates ‘max_mismatch_bp’ was set to 1, and the mapping quality threshold was set to 30. Filtered read pairs were aggregated into genomic bins of different sizes using the cooler (v.0.8.11)77. The resulting Hi-C matrices were normalized using the iterative correction procedure.
Compartment annotation
A and B compartments were annotated using the cooltools (v.0.3.2) call-compartments function for 200 kb resolution contact matrices. The orientation of the eigenvectors (PC1) was selected such that it correlates positively with GC content and expression data. Consequently, B compartment bins were assigned with negative eigenvector values, and A compartment bins were assigned with positive.
Loops and TADs annotation
High-resolution Hi-C data for HAP1 cells22 at 10 kb resolution were used for loops and TADs annotation. Loops were annotated using Chromosight (v.1.4.1)78. For loop detection, the Pearson correlation threshold was set to 0.4, loop sizes were set between 50 kb and 5 Mb and the parameter ‘–smooth-trend’ was enabled. TADs were annotated using the insulation score algorithm implemented in the cooltools (v.0.3.2) diamond-insulation function79. The window size for insulation score calculations was set to 200 kb. The threshold for the boundary strength filter was calculated using the Li method, implemented in the scikit-image package80. The bins with boundary strength higher than ~0.19 were considered as TAD boundary bins. These bins were converted into TADs by continuously joining two neighboring bins together. The TAD boundary coordinate was then randomly selected from the coordinates of the joined bins with a significant insulation score.
Aggregate analyses
Average loops, TAD boundaries and TADs were calculated for 10 kb resolution observed-over-expected Hi-C contact matrices using the loops and TADs annotated as described above. Publicly available HAP1 Hi-C data were included for comparison6,22. Expected contact matrices were obtained using the cooltools (v.0.3.2) function ‘compute-expected’79. Average loops were generated using coolpup.py (v.0.9.5)81 with ‘pad’ set to 200 and ‘min-dist’ set to 0. Average TAD boundaries were generated using coolpup.py (v.0.9.5)81 in ‘local’ mode with ‘pad’ set to 500. Average TADs generated using coolpup.py (v.0.9.5)81 in ‘local’ mode with the ‘rescale’ option, with the ‘rescale_size’ set to 99. The average loop strength was calculated as the mean value of the central three-by-three square pixels. The average TAD boundary strength was calculated as the mean value of the average intra-TAD interactions (upper-left and bottom-right quarters) divided by the mean value of average inter-TAD interactions (upper-right and bottom-left quarters). The average TAD density was calculated as the mean value of the central 33-by-33 square pixels.
The aggregate stripes analysis of the TetO integrations was performed using cooltools (v.0.5.1)79 and bioframe (v.0.3.0)82 for 10 kb resolution observed-over-expected Hi-C contact matrices. The pile-ups of the TetO integrations were created using the cooltools.pileup function with 500 kb regions around the integration coordinates as flanks.
Statistics and reproducibility
All comparisons were made between biologically independent samples. No statistical method was used to predetermine sample size. No data were excluded from the analyses. The experiments were not randomized. The Investigators were not blinded to allocation during experiments and outcome assessment. Data distribution was assumed to be normal, but this was not formally tested.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.