Samples
This study included 119 C. burnetii-positive samples that had been previously confirmed using molecular methods, including nested PCR and TaqMan real-time PCR assays. The samples were obtained from a wide range of sources, both human and animal, and were collected from different regions across Iran. Animal-derived specimens comprised abortion materials and milk samples from cattle, sheep, and goats, whereas the human sample was a clinical specimen obtained from a patient diagnosed with Q fever endocarditis. To evaluate the suitability of these samples for genotyping, all 119 specimens were re-screened for the presence of the C. burnetii IS1111 gene using TaqMan real-time PCR assay [11,12,13,14]. Based on the quantification cycle (Ct) values, only samples with Ct ≤ 32, reflecting sufficient bacterial DNA quantity and quality, were selected for MLVA. According to this criterion, 26 samples were deemed appropriate for the typing. The dataset included a combination of bovine, ovine, and caprine abortion samples, milk from cows, sheep, and goats, and one human heart valve specimen. The final set used for MLVA genotyping consisted of 25 animal samples and one human clinical sample, as detailed in Table 1 [11,12,13,14].
Table 1 Number of samples selected for C. burnetii genotypingVNTR PCR amplification
PCR amplification was conducted to target nine loci of C. burnetii (ms01, ms03, ms07, ms12, ms21, ms22, ms26, ms30, ms31, and ms36) for genotyping via gel electrophoresis. Additionally, six loci (ms23, ms24, ms27, ms28, ms33, and ms34) were amplified for genotyping by capillary electrophoresis [10,11,12,13,14,15]. Primer sequences for all loci were aligned with the MLVA database for C. burnetii (Table 2). Specifically, the reverse primers for loci ms23, ms33, and ms34 were labelled with the fluorescent dye 6-FAM, whereas those for ms24, ms27, and ms28 were labelled with 6-carboxy 4,7,2’,4’,5’,7’ hexachloro fluorescein (HEX) [13]. Each PCR reaction was prepared in a total volume of 25 µL, comprising 5 µL of extracted DNA, 12.5 µL of TEMPase Hot Star 2x Master Mix BLUE (Amplicon, Odense M, Denmark), and 400 nM of each primer. Sterile distilled water was added to bring the total volume up to 25 µL. The PCR cycling protocol consisted of an initial denaturation step at 95 °C for 15 min, followed by 40 cycles of denaturation at 94 °C for 30 s, locus-specific annealing at optimized temperatures for 60 s, and extension at 72 °C for 1 min. The final extension step was performed at 72 °C for 10 min to ensure complete amplification of the target loci. The Nine-Mile RSA 493 strain of C. burnetii was used as the reference strain, following established guidelines. To validate the specificity of amplification, sterile distilled water served as a negative control throughout the PCR process, thereby ensuring the reliability and accuracy of the results.
Gel electrophoresis of PCR products
The PCR products corresponding to each locus, including ms01, ms03, ms07, ms12, ms21, ms22, ms26, ms30, ms31, and ms36, were resolved on agarose gels with varying concentrations tailored to the estimated fragment sizes [10]. Specifically, 1% agarose was used for loci ms07 and ms12, 1.5% agarose for locus ms36, 2% agarose for loci ms01, ms03, ms21, ms22, ms30, and ms31, and 2.5% agarose for locus ms26. Following electrophoresis, bands corresponding to the PCR products were visualized using a gel documentation imaging system under ultraviolet (UV) light. In cases where no PCR product band was detected, the amplification process was repeated for the affected isolate. If repeated amplifications failed to yield a product, the corresponding allele was designated as null. Fragment sizes were subsequently analyzed using the Gel Analyzer software (SequentiX) to ensure accurate measurement and characterization of the PCR products.
Capillary electrophoresis of fluorescently labelled PCR products
Following the amplification of the six loci, capillary electrophoresis was performed using the Applied Biosystems Genetic Analyzer model 3130x (UK), renowned for its high precision in fragment sizing. The PCR products were purified using the Kowsar PCR Product Purification Kit, and each sample was then diluted at ratios of 1:5, 1:10, and 1:100. For the analysis, 2 µL of each diluted sample was mixed with 10.5 µL of formamide and 0.5 µL of a 560 bp standard size marker (ROX, Applied Biosystems). This mixture was thoroughly mixed, denatured at 95 °C for 30 s, and placed on ice for 2 min to halt the reaction. The prepared samples were subsequently analyzed in an AB3130XL capillary sequencer using the POP7 polymer at the Pasteur Institute of Iran. The data obtained from capillary electrophoresis were processed using Gene Marker software (version 1.6), focusing on the sizes of the standard markers and corresponding fluorescent signals associated with each locus. The 6-FAM dye emitted a blue signal, whereas the HEX dye emitted a green signal. This differentiation enabled the precise sizing of the fragments, which were identified and recorded based on the peaks generated during electrophoresis.
VNTR data analysis
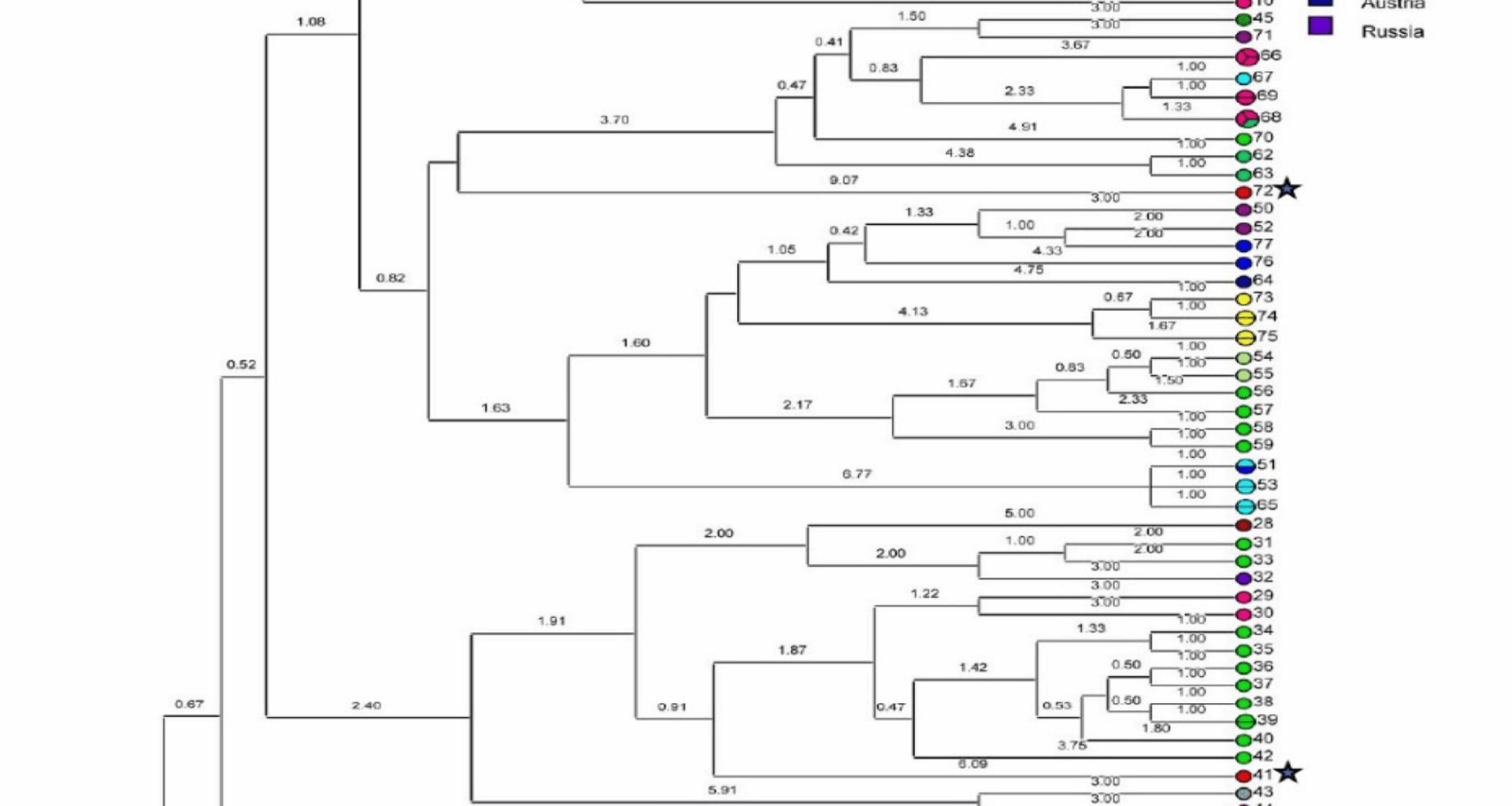
The fragment sizes obtained for each sample were analyzed to determine the number of repeats at each VNTR locus. The repeat counts for each VNTR locus were calculated using fragment sizes and repeat numbers from the reference strain Nine Mile RSA493 of C. burnetii [16]. The calculation of repeat numbers was based on the following equation: number of repeats (bp) = (fragment size (bp) − flanking regions (bp))/repeat size (bp) [17]. The MLVA data from the collected samples were then analyzed using the online database at http://microbesgenotyping.i2bc.paris-saclay.fr. Each sample’s profile was compared against database profiles, with unmatched profiles classified as novel C. burnetii genotypes. Data from the online database were further analyzed using Bionumerics 6.6 software (Applied Maths, Sint-Martens-Latem, Belgium), in which both the study data and database entries were examined using bioinformatics tools across 15 loci. To compare C. burnetii strains, the Unweighted Pair Group Method (UPGMA) was applied. Cluster analysis was conducted using MLVA/VNTR gel electrophoresis for genomic grouping. The discriminatory power of the MLVA genotyping method was evaluated using Simpson’s diversity index, as per Hunter and Gaston [18], calculated for each locus, each spacer, and the entire MLVA dataset to assess the ability of the technique to effectively differentiate and classify genotypes.
Ethical code
This study, including the proposal, all experimental protocols, and consent procedure, was approved by the Ethics Committee for Biomedical Research of Tarbiat Modares University (Ethic Code: IR.TMU.REC.1395.510). All methods were carried out following the relevant guidelines and regulations of the Ethics Committee for Biomedical Research of Tarbiat Modares University. The related methods are reported following the ARRIVE guidelines (https://arriveguidelines.org).