09 Jan 2026
When TDP-43 escapes from the nucleus, cells bungle mRNA processing, chopping up and splicing transcripts in the wrong places. Most studies have focused on retention of “cryptic” exons, but recent work has shone a spotlight on a different aberration: misplaced polyA tails. Do these errors affect disease? In the January 5 PloS Biology, scientists led by Leonard Petrucelli at the Mayo Clinic in Jacksonville, Florida, and Aaron Gitler at Stanford University School of Medicine suggested they do.
- In FTLD-TDP brain, VPS35 and ELK1 transcripts were shifted toward longer forms.
- Both changes correlated with markers of TDP-43 pathology.
- The amount of VPS35 protein was halved, disrupting retromers.
In postmortem frontal cortex sections from more than 200 people with frontotemporal lobar degeneration due to TDP-43, the authors linked alternative polyadenylation (APA) of the retromer gene VPS35 to TDP-43 pathology. Abnormal VPS35 transcripts slashed the amount of VPS35 protein, a key component of the retromer complex. Because retromers ferry proteases to the lysosome, their loss would cripple the cell’s ability to handle waste. In addition, transcription factor ELK1 underwent APA that correlated not only with markers of pathology, but with earlier disease onset as well.
“This is the first clear evidence that TDP-43-driven alterations in alternative polyadenylation translate into meaningful biological consequences at the protein level in the FTLD-TDP brain,” co-first author Vidhya Maheswari Jawahar at Mayo wrote to Alzforum. “This work establishes APA as a disease-relevant mechanism and potential therapeutic target in TDP-43 proteinopathies.”
Steven Finkbeiner at the University of California, San Francisco, noted that VPS35 has also been linked to Alzheimer’s disease, while ELK1 regulates genes involved in synaptic plasticity. “Both VPS35 and ELK1 are potentially relevant for neurodegeneration and memory loss,” he wrote to Alzforum (comment below).
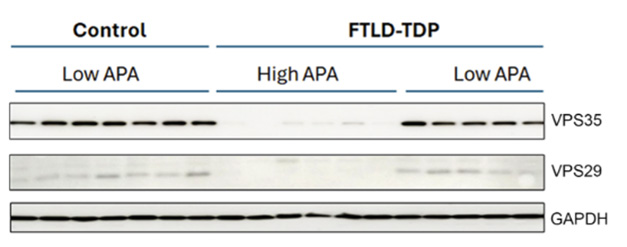
Poof! In FTLD-TDP brains with rampant alternative polyadenylation of the VPS35 transcript, retromer component VPS35 appears to have vanished and VPS29 is barely detectable compared with their levels in low-APA FTLD and control brain. Loading control GAPDH is unaffected. [Courtesy of Maheswari Jawahar et al., PLoS Biology, 2025.]
TDP-43’s role in suppressing cryptic exons was first reported a decade ago, and early studies identified the microtubule regulator stathmin-2 as one of the most affected transcripts (Aug 2015 news; Jan 2019 news).
More recently, scientists have homed in on misplaced tails. In a previous collaboration, Gitler and Petrucelli sequenced the 3′ end of transcripts in iPSC-derived neurons (iNeurons) that lacked TDP-43 to search for APA events. They found more than 400 genes where APA boosted or squelched gene expression, hinting at biological effects. Two other groups reported similar APA events at the same time (Oct 2025 news). Still, it was unclear whether these changes affect disease.
To find out, Maheswari Jawahar and co-first author Yi Zeng at Stanford followed up on three of the strongest hits, ELK1 and the retromer proteins VPS35 and VPS26B. They examined frontal cortex sections from 220 FTLD-TDP cases and 51 healthy controls. At the time of death, the FTLD-TDP patients were 73 years old on average and had had symptoms for eight years. A quarter had motor neuron degeneration, a sign of amyotrophic lateral sclerosis.
The authors extracted bulk RNA from these sections and ran real-time PCR to quantify APA and total transcripts. VPS26B transcripts were not significantly different in FTLD and control brain, and the authors did not study this gene further. By contrast, ELK1 and VPS35 transcripts were shifted toward longer isoforms in FTLD patients, as they had been in iNeurons. Longer transcripts crop up when the adenylation machinery fastens the polyA tail too far downstream, lengthening the 3′ untranslated region.
The extra 3′ UTR material had opposing effects on the two genes. For ELK1, the additional nucleotides stabilized the transcript and boosted translation. Conversely, VPS35 protein levels were slashed in half. By western blot, this effect was striking, with VPS35 protein nearly undetectable in the half of the cohort that had the highest ratio of long to total transcripts (image above). VPS35 is the central hub of the retromer complex, and its loss typically leads to degradation of other components. In keeping with this, the amount of VPS29 protein plummeted in tandem with VPS35 in FTLD brains, with a correlation coefficient of 0.78.
Co-author Scott Small at Columbia University in New York, a retromer biologist, noted that many cellular alterations can disrupt retromers, but few do so to this degree. “From our model system work, there is no question that this degree of deficiency will functionally disrupt retromer-dependent endosomal recycling, causing endolysosomal network dysfunction,” he wrote to Alzforum.
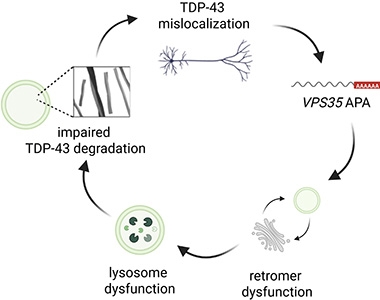
Vicious Cycle? Loss of nuclear TDP-43 (top) triggers misplacement of the polyA tail on VPS35 transcripts (right), leading to impaired retromers and lysosomes (bottom), which in turn causes further TDP-43 buildup (left). [Courtesy of Maheswari Jawahar et al., PLoS Biology, 2025.]
Notably, a prior study had reported that knocking out VPS35 in a mouse led to TDP-43 accumulation and neurodegeneration (Tang et al., 2020). Similarly, Petrucelli and colleagues previously found that handicapping endosomes caused TDP-43 to aggregate (Shao et al., 2022). Together, the data suggest a model in which loss of nuclear TDP-43 knocks down VPS35, and loss of VPS35 further exacerbates TDP-43 pathology in a vicious cycle, the authors proposed (image at right).
In keeping with this, in FTLD-TDP cortex, VPS35 APA correlated with having more cryptic stathmin-2, a marker of pathology. Intriguingly, ELK1 APA was even more tightly linked to disease. It correlated with having more phosphorylated TDP-43 as well as more cryptic stathmin-2, and with a younger age of disease onset. All of these relationships were weak, with most having a correlation coefficient around 0.2, but were statistically significant. Maheswari Jawahar noted that weak correlations are to be expected in a complex disease such as FTLD, particularly when using bulk RNA analysis.
Commenters cautioned that correlations do not equal causation. “It’s unclear to what extent these changes actually contribute to FTD pathogenesis,” Finkbeiner wrote. Sami Barmada at the University of Michigan, Ann Arbor, wondered if these APA events promote neurodegeneration by themselves, or simply reflect TDP-43 dysfunction (comment below). However, both scientists thought the APA changes have potential as biomarkers for TDP-43 proteinopathy.
Maheswari Jawahar said that she will next try to determine how APA affects endolysosomes and pathology. She also plans to pin down cell-type-specific consequences using single-cell RNA-Seq. If APA does drive pathology, that would hint at therapeutic approaches, such as stabilizing retromers or boosting endolysosome function, she said.
Huda Zoghbi at Baylor College of Medicine, Houston, agreed that enhancing lysosomal function could help ameliorate pathology, but she suggested the most effective way to intervene in TDP-43 proteinopathies would be to keep TDP-43 nuclear (comment below).
Meanwhile, Peter Nelson at the University of Kentucky, Lexington, wondered about other neurodegenerative diseases that accumulate TDP-43 yet have distinct characteristics. “As someone who studies the most common subtype of TDP-43 proteinopathy, LATE-NC, I wonder what the polyadenylation/VPS35/ELK1 picture is for that condition, and if this molecular heterogeneity provides an added basis for disease classification,” he wrote to Alzforum (comment below).
This paper is part of a surge in interest in TDP-43 biology. Scientists at the University of Wisconsin-Madison have put out a call for abstracts for a TDP-43 biomarker conference to be held May 21 in Madison.—Madolyn Bowman Rogers
News Citations
- Does New Role for ALS-Linked Protein Help Explain Neurodegeneration? 8 Aug 2015
- Microtubule Regulator Connects TDP-43 to Axonal Dysfunction 18 Jan 2019
- When TDP-43 Ghosts the Nucleus, Cryptic Poly-A Sites Rise from the Dead 31 Oct 2025
Paper Citations
-
Tang FL, Zhao L, Zhao Y, Sun D, Zhu XJ, Mei L, Xiong WC.
Coupling of terminal differentiation deficit with neurodegenerative pathology in Vps35-deficient pyramidal neurons.
Cell Death Differ. 2020 Jul;27(7):2099-2116. Epub 2020 Jan 6
PubMed. -
Shao W, Todd TW, Wu Y, Jones CY, Tong J, Jansen-West K, Daughrity LM, Park J, Koike Y, Kurti A, Yue M, Castanedes-Casey M, Del Rosso G, Dunmore JA, Zanetti Alepuz D, Oskarsson B, Dickson DW, Cook CN, Prudencio M, Gendron TF, Fryer JD, Zhang YJ, Petrucelli L.
Two FTD-ALS genes converge on the endosomal pathway to induce TDP-43 pathology and degeneration.
Science. 2022 Oct 7;378(6615):94-99. Epub 2022 Oct 6
PubMed.
External Citations