VP2 mutation analysis
Comparison of the amino acid sequences of the CPV-positive strains with those of CPV2 Pfizer (vaccine)−06 revealed multiple amino acid mutations; the feline-derived CPV-GD07-23 carried a P495H mutation, CPV-GD06-24 carried an S27Y mutation, CPV2c-GD05-23 carried Q318P, N323H, and N505K mutations, and CPV2c-GD03-24 carried an A537P mutation were identified for the first time, the feline-derived CPV2c-GD08-23 and canine-derived CPV2c-GD01-23, CPV2c-GD09-23 were found to have A5G mutations. We conducted a detailed sequence alignment between the A5G-positive strains in this study and the earliest reported A5G-containing CPV-2c genomes in China, including 18 strains reported by Wang et al. [25] in 2016 (all with gene sequences identical to accession numbers KT162005.1/KT162021.1) and subsequent isolates cited in reference [26] (accession numbers KY937650/KY937662). The main findings are as follows: The VP2 sequences of the A5G strains in this study share 98.2%−100% identity with the 2016 A5G strains. Additionally, the A5G mutation is stably retained in the same genetic context, with the flanking sequences at positions 1–10 of VP2 being 100% conserved. In the updated phylogenetic tree (Fig. 2), all A5G-positive strains—including those from this study and earlier isolates—form an independent subclade within the broader CPV-2c branch. This confirms their strains with a common evolutionary ancestor and supports the existence of the “CPV-2c A5G subvariant.” These direct comparison results further verify that A5G is not a sporadic mutation but a signature conserved feature of the emerging CPV-2c subvariant. In this study, all isolate strains were amplified using PCR with the full-length VP2 primers VP2-F/R. These sequences were compared and analysed with the NCBI reference sequences, which identified fourteen strains of FPV and nineteen of CPV. The amino acid sequences of the FPV-positive strains were compared with those of FPV-Italian Pfizer (vaccine)−08, which revealed mutations at positions 31, 300, 562, 568. Thirteen CPV strains collected in this study were identified as CPV-2c, which is more common than other variants and has a shared evolutionary origin with Asian strains [27]. An analysis of 1,076 VP2 sequences of Chinese CPV-2 strains from the NCBI database revealed a clear upward trend in the prevalence of CPV-2c: CPV-2c was first reported in China in 2009, with an initially extremely low proportion; Between 2017 and 2018, the proportion of CPV-2c surpassed that of CPV-2a, becoming the dominant subtype nationwide [26]. Literature data show that CPV-2c in Guangdong rose from a minor subtype to a dominant subtype between 2016 and 2019 [26]. In our study (2023–2024), CPV-2c accounted for 64.7% of canine-derived CPV isolates in Guangdong, with stable detection of 2 feline-derived CPV-2c strains, While our sample collection period was brief and the sample size relatively small, a comprehensive analysis of the aforementioned literature data still confirms, to a certain extent, the ongoing transmission of CPV-2c in Guangdong [26,27,28,29].
Feline-derived CPV-2c identification, evolution and cross-species transmission
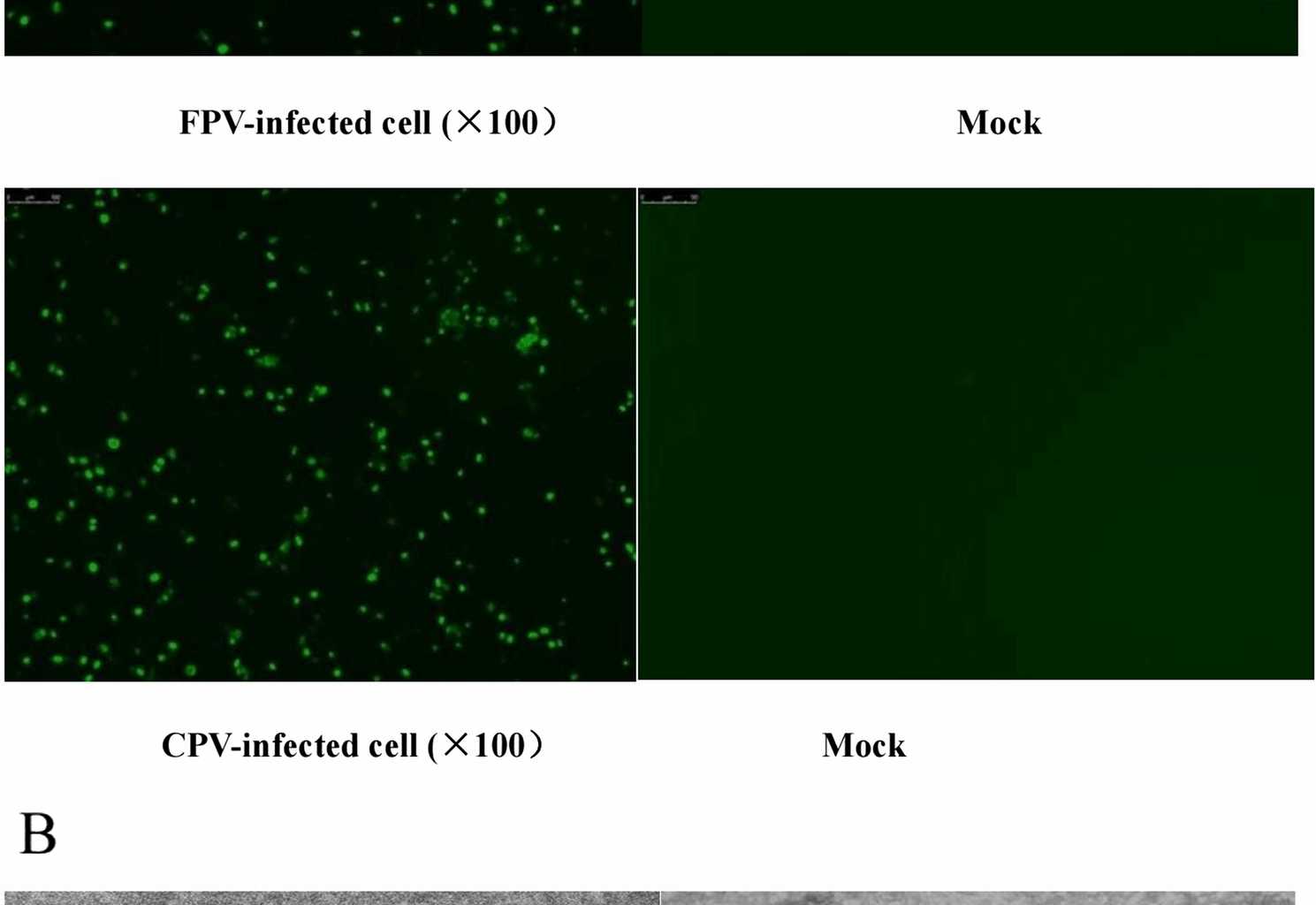
Initially, two cases of suspected feline-derived parvovirus infection collected from a pet hospital were identified using PCR. However, after amplification and sequencing of the complete sequence of VP2 and comparison with the amino acid sequences of FPV-CU-4 and FPV-Italy-Pfizer (vaccine)−08, we found that positions 80, 93, 103, 300, 323, 564, and 568 were inconsistent with each other and that the homology with CPV was much higher. Further, by comparing the amino acid sequence with that of CPV2-Pfizer (vaccine)−06, the mutations S297A and N426E were identified, therefore, it was finally identified as CPV-2c [30,31,32]. Two amino acid changes in VP2 on the surface of CPV can lead to a host switch from dogs to cats because these mutations are concentrated in the binding domain of VP2, which binds to the transferrin receptor of the host cell, and thus affect the binding of the viral particle to the the receptor for transferrin, which is closely associated with CPV infection [32, 33].
Building on our phylogenetic tree (Fig. 2) constructed using VP2 gene sequences, we highlighted the clustering relationship between feline- and canine-derived strains through color labeling (e.g., feline-derived CPV-2c in red and canine-derived CPV-2c in blue). This visualization reveals a close genetic relationship between local feline- and canine-derived CPV isolates: the two feline-derived CPV-2c strains (CPV2c-GD07-23 and CPV2c-GD08-23) share a high degree of similarity in VP2 gene sequences with canine-derived CPV-2c strains—exhibiting over 99% homology with CPV-2c isolates or reference strains, while their homology with FPV isolates or reference strains ranges from 97–98%—and cluster within the same evolutionary branch, supporting a close genetic relationship between them. This clustering pattern indicates that they share a recent common ancestor with local canine CPV-2c, rather than forming an independent, distantly related branch. This phylogenetic pattern supports the hypothesis that local dog populations serve as the reservoir for CPV, with occasional spillover transmission to cats, facilitating viral persistence through a “host-switching cycle” [17, 18, 34]: The nesting of feline CPV-2c within the canine CPV-2c clade suggests that feline infections are not sporadic events or introduced from external sources, but rather result from ongoing cross-species transmission from local dog populations. This aligns with our sequence alignment observations, where feline and canine strains share conserved genetic motifs (e.g., conserved regions in VP2); Unique mutations in feline isolates (such as A5G and P495H in VP2, Table 4) likely emerged post-spillover, representing adaptive changes to optimize replication in feline hosts. Despite their host specificity, these mutations do not disrupt the overall phylogenetic affinity with canine strains, indicating that feline CPV remains genetically linked to its canine counterparts. This host-associated phylogenetic structure reveals a key persistence strategy of CPV: the virus primarily maintains itself in dog populations (as the dominant reservoir) while utilizing cats as a secondary host. This dual-host dynamic reduces the risk of viral extinction due to fluctuations in a single host population (e.g., seasonal changes in dog density) and enables continuous genetic exchange between host-adapted variants.
A review of clinical records for these cases showed that the symptoms of CPV-2c-infected cats were similar to those of typical FPV infection (feline panleukopenia) but with overall milder severity. No concurrent FPV infection was detected in these CPV-2c-positive cats, as samples were pre-screened by multiplex PCR to exclude co-infections with FPV, FCV, and other viruses prior to virus isolation. We speculate that these differences in symptom severity may be related to the adaptive stage of CPV-2c in cross-species transmission. As a strain transmitted from dogs to cats, its affinity for feline host cell receptors and replication efficiency may not yet be fully optimized, resulting in slightly lower pathogenicity compared to FPV, which is highly adapted to feline hosts.
In this study, feline-derived CPV rather than canine-derived CPV was selected for experiments, aiming to explore different evolutionary stages of viral host adaptation. Unlike studies on canine-derived CPV, which mainly focus on the initial process of the virus breaking through cross-species barriers, our research centers on the “post-adaptation stabilization period”. The CPV-2c-GD08-23 strain, isolated from a naturally infected cat with clinical symptoms, has developed basic adaptability to feline hosts. Through serial passage in CRFK cells, we were able to observe: whether the adaptive mutations acquired in natural infections are stably retained under long-term selection pressure from feline cells; whether the emergence of reversion mutations (such as G568A) reflects evolutionary constraints (for example, certain mutations are structurally incompatible with the feline cellular environment); the association between genetic changes and replication adaptability (such as the decrease in titer after reversion mutations). This helps to clarify the “stability of cross-species adaptation”—a key yet understudied aspect in the evolution of viral host range. It is worth noting that this study complements rather than replaces research on canine-derived CPV: studies on canine-derived CPV answer “how CPV replicates in feline cells for the first time”, while our research answers “how established feline-derived CPV maintains replication”. Together, they form a comprehensive understanding of CPV host adaptation. Future studies will include canine-derived CPV strains to verify their adaptability to feline cells and feline hosts, thereby combining the dynamics of the initial cross-species stage and the post-adaptation stage to construct a complete picture of CPV evolution.
Evolution and recombination
Viral genetic variation is a fundamental driver of evolution [35, 36], and our data reveal distinct evolutionary patterns between FPV and CPV that align with and extend prior insights [37]. FPV exhibits striking sequence conservation: in our isolates, VP2 gene nucleotide variations were detected, but most were synonymous mutations—leaving the VP2 protein’s amino acid sequence unchanged. This confirms FPV’s stability at the protein level, consistent with previous observations. In contrast, CPV shows dynamic VP2 evolution: our isolates display abundant non-synonymous mutations (e.g., Gly31Ala and Pro495His in feline CPV-2c; Glu318Pro in canine CPV) that directly alter VP2 structure, validating that CPV’s VP2 evolves in tandem with genetic changes.Notably, minor genetic alterations in CPV drive significant functional impacts. Despite small numbers of nucleotide changes per mutation (mirroring the pattern where CPV-2 evolved into 2a via 5 amino acid mutations, and further into 2b/2c with fewer changes), structural modeling confirms tangible effects: for example, Pro495His disrupts antigenic sites, while Ala300Gly modulates hydrophobic interactions—indicating these are adaptive, not neutral, changes, and underscoring how small variations can reshape antigenicity and pathogenicity. Consistent with reports [38,39,40], CPV evolves far faster than FPV: the accumulation of non-synonymous VP2 mutations in CPV (but not FPV) in our samples reflects its heightened evolutionary dynamism, with VP2 evolving at a rate (~ 10⁻⁴ substitutions/site/year) comparable to some RNA viruses [41]. In essence, our findings anchor empirical data in evolutionary theory: FPV’s conserved VP2 (synonymous mutations) contrasts with CPV’s variable VP2 (functional amino acid changes), while the functional potency of CPV’s small genetic alterations highlights its unique adaptive potential—key to understanding its epidemiological success.
Although the genetic evolution of FPV and CPV has been extensively studied, the recombination, co-circulation, and evolution of FPV and CPV have rarely been addressed. In the present study, FPV and CPV recombination analyses were performed on the positive strains, and a total of four possible FPV and CPV recombination evolutionary events were found. These findings suggest that FPV and CPV are not only capable of undergoing independent genetic mutations within hosts but may also undergo recombinant evolution through the exchange of genetic materials [42]– [43].
Structural interpretation
Based on PyMol structural analysis and passage experiments, our conclusions are: Some mutations are directly related to adjustments in viral virulence or replication capacity, while others primarily affect antigenicity rather than virulence. “Mutations affecting antigenicity but not direct virulence” include mutations such as A300G in FPV and P495H in feline-derived CPV. Structural analysis showed that they primarily alter the antigenic index or hydrophobic regions of the VP2 protein (significantly different from vaccine strains) but are not directly associated with changes in virulence (e.g., no decrease in viral titer). The core risk of these mutations is their potential to reduce the recognition efficiency of antibodies induced by existing vaccines, rather than enhancing virulence. Specifically, the FPV VP2 A300G (Ala→Gly) mutation introduces glycine (a side chain-free amino acid) and disrupts hydrogen bonds, leading to increased backbone flexibility. This enhanced structural plasticity may improve receptor-binding adaptability, potentially facilitating viral adaptation to different hosts. Mutagenesis studies by Truyen et al. [43] have shown that substitutions at residue 300 can relax the constraints on transferrin receptor (TfR) binding, enabling CPV to infect non-canonical hosts. Research has demonstrated that alterations at position 300 may modify host range and synergistically coordinate with other mutation sites to alter viral binding efficiency with the transferrin receptor (TfR), thereby influencing infection efficiency [44]. Studies indicate that the 300th amino acid residue constitutes a critical position within antigenic site B, which may be associated with neutralizing antibody production [45]. Similar to the A300G mutation reported by Parrish et al. [46], mutations at residue 300 alter the conformational flexibility of antigenic site B, directly reducing the binding efficiency of vaccine-induced neutralizing antibodies. In the present study, the A300G mutation in feline-derived FPV isolates may play an important role in enhancing the flexibility of the VP2 protein and thereby influencing viral replication. The CPV2c VP2 P495H (Pro→His) mutation alters the tertiary structure by replacing the rigid pyrrolidine ring of proline with histidine, resulting in a more flexible loop conformation. The adjacent beta-turn is located near the capsid assembly interface, and the reduced structural stability may impair viral particle assembly efficiency. To date, the functional implications of this mutation have not been reported in the literature and warrant further investigation.
“Mutations associated with reduced virulence” refer to the G568A reversion mutation in feline-derived CPV-2c (appearing at passage 16): Structural predictions showed that this mutation restores the rigid conformation of the VP2 protein. Simultaneously, a decrease in viral titer was observed in passage experiments of the CPV2c-GD08-23 strain, where this reversion—changing from glycine back to alanine in the wild-type virus—coincided with reduced virulence, as attenuated cytopathic effects were observed in cells compared to earlier passages. This suggests that the mutation may achieve “adaptive attenuation” in feline cells by reducing replication efficiency (avoiding excessive damage to host cells, which is beneficial for sustained viral persistence), though the underlying mechanisms for this attenuation require further investigation. Notably, the FPV VP2 A568G (Ala→Gly) mutation, conversely, induces tertiary structural changes that may alter viral replication dynamics in the host. Crystallographic studies have shown that residue 568 is located in the β-sheet region of the VP2 protein, adjacent to the hydrophobic core inside the capsid and the pentamer interaction interface [47, 48]. The amino acid composition of this region is crucial for maintaining capsid stability and viral particle assembly efficiency; for instance, structural analysis by Parrish’s team (2003) revealed that conservative mutations in this region can affect the viral replication cycle in host cells by altering the hydrogen bond network inside the capsid [47]. Studies [17, 49] have further identified the 568th amino acid residue as critical for efficient viral replication in the host, which aligns with our observation in cell passage experiments: the viral titer decreased in the 16th passage due to the G568A reversion mutation, suggesting that this residue may influence replication fitness through structural regulation. Our data suggest “CPV-2c may have a tendency of reduced pathogenicity in CRFK cells due to gene mutations” but cannot confirm pathogenicity decreases with spread. Direct experimental evidence is limited to the G568A mutation in one strain, with reduced titer in feline cells; potential structure-function links lack definitive pathogenicity correlates. Key limitations include small sample size, absence of longitudinal clinical data and the need to verify single-mutation impacts via reverse genetics-derived mutants in cat challenge experiments.
This study does not claim all mutations are significant, but rather screens for candidate functional mutations related to host adaptation and antigenicity through structural modeling and passage experiments—with unvalidated mutations explicitly requiring further research. Its novelty lies in analyzing “structure-function-host adaptation” associations of multiple key mutations, offering a new perspective on CPV-2c’s evolutionary mechanisms. Cell-level functional experiments (e.g., reverse genetics-based mutant construction) are planned to supplement these findings.
Clinical and preventive implications
In this study, by analyzing the VP2 amino acid differences between the genotypes of the isolated strains and commercial vaccine strains, it is speculated that the antigenic differences from commercial vaccine strains may lead to insufficient cross-protection of existing vaccines. This aligns with the fact that current feline panleukopenia vaccines, which are based on FPV strains, have no definitive clinical data confirming their protective efficacy against feline-derived CPV-2c. However, existing studies suggest that FPV and CPV share certain antigenic cross-reactivity—particularly in conserved regions of the VP2 protein—implying that current vaccines may provide partial cross-protection, albeit likely weaker than the homologous protection against FPV. This reduced efficacy is presumably linked to VP2 amino acid differences, mutations such as S297A and N426E, which have been confirmed in previous studies, which may impair antigen recognition. Although there is currently no direct clinical failure data, Smith et al. clearly proposed in a 2025 study published in Vaccine that the antigenic matching degree between vaccine strains and circulating strains is a key factor determining cross-protection efficacy. Cavalli A, Martella V, et al. [50] confirmed through antigenic cross-reactivity assays that cumulative genotypic differences—such as mutations at key VP2 sites—between various canine parvovirus type 2 (CPV-2) variants (particularly CPV-2c) and the original strain, as well as among the variants themselves, have resulted in significant antigenic divergence. These findings, coupled with evidence of reduced cross-neutralization efficacy of antibodies induced by existing vaccines against emerging variants like CPV-2c, suggest that regular updating of vaccine strains to match circulating genotypes is a necessary measure to sustain the effectiveness of prevention and control efforts.
Chen S, et al. [51] pointed out that canine parvovirus type 2 (CPV-2) variants can spread from felids and canids to species of Carnivora (such as tigers and lions), Artiodactyla (such as pigs), and Pholidota (such as pangolins). Among them, CPV-2c is a key variant for transmission from Carnivora to Artiodactyla and Pholidota. In this study, we detected rich genotypic diversity in animal samples, including 2 strains of CPV-2c in cats. The increasing frequency of contact between domestic pets and wild animals such as stray cats and raccoon dogs creates conditions for viral recombination and cross-species transmission. As highlighted in a 2024 study in Science of the Total Environment [51], the persistent risk of cross-species transmission necessitates continuous surveillance. As CPV-2c adapts and evolves in feline populations, its antigenic epitopes may undergo further mutations. Thus, long-term epidemiological investigations combined with neutralization tests are required to dynamically evaluate the protective efficacy of existing vaccines against variant strains, ensuring that vaccination strategies can address viral adaptive changes. Strengthening surveillance at the human-animal interface—especially in areas with frequent contact between domestic pets and wild animals—is also crucial for timely detection and prevention of potential zoonotic risks.
Study limitations and outlook
This study has several limitations Firstly, the epidemiological analyses were constrained by a relatively small sample size, which may reduce the statistical power to detect significant associations. The samples in this study were only collected from hospital-referred animals (mainly pets with clinical symptoms), which may not fully represent healthy populations or stray animal populations (the latter often lack medical records), indicating a potential selection bias. In future studies, we will adopt stratified sampling (covering household pets, stray animals, and farm-raised populations) to address this limitation. Secondly, the sampling methodology presents geographical and ecological constraints – all specimens were exclusively obtained from veterinary hospitals within Guangdong Province. This restricted sampling framework consequently limits the extrapolation of our results to the broader South China region. Thirdly, Lack of Functional Validation: Structural predictions of VP2 mutations were not experimentally confirmed via reverse genetics or neutralization assays.
Despite certain limitations in this study, it still holds value beyond mere observations and includes some significance in exploring mechanisms. The core findings can be summarized into three interconnected aspects: novel evolutionary changes (mutations/recombination) drive the adaptation of canine parvovirus (CPV) to feline hosts, thereby increasing its epidemiological risk in cat populations. The core insights are summarized as follows: Viral evolution: Unique mutations (e.g., Gly31Ala and Ala568Gly in FPV, Pro495His in feline-derived CPV-2c) were identified in FPV as well as feline-derived CPV in southern China. Structural modeling confirmed that these mutations affect VP2 stability, indicating they are adaptive evolutionary changes rather than random variations; CPV risk in cat populations: Cats in southern China are not transient hosts of CPV but potential reservoirs. High-load feline-derived CPV strains were isolated from positive samples, demonstrating stable viral replication in cats; Host adaptation: Serial passage experiments of the feline-derived CPV-2c strain revealed that mutations (G568A reversion mutation) correlate with replication efficiency. Additionally, the feline isolate-specific P495H mutation (in the host receptor-binding domain) provides direct evidence that CPV adapts to feline cells through mutations, challenging the traditional view that CPV persists only transiently in cats.
Subsequent studies will systematically advance the following work: For key mutations, in vitro neutralization assays are planned, where vaccine-immune sera will be incubated with mutant strains to verify their impact on antibody binding efficiency. Meanwhile, infection experiments will be conducted using primary intestinal epithelial cells from canines and felines; through the TCID₅₀ assay, the replication efficiency of mutant and wild-type strains will be compared to evaluate differences in their host adaptation capacity. Building on these efforts, future research will further focus on large-scale sampling across ecological gradients and functional validation of mutations, aiming to translate current findings into actionable intervention strategies and provide practical scientific basis for viral prevention and control.