The present work was undertaken to use WGS to detect MRMP, p1 type, MLST, as well as to perform core-genome analysis. The study was run on a group of 38 M. pneumoniae strains isolated in the UK during 2016–2024. The strains were selected randomly to assess the viability of the WGS pipeline and to test the efficiency of the method. Core-genome analysis was used to compare the 38 M. pneumoniae UK data with the global genomic data of 290 M. pneumoniae strains from 14 countries isolated between 1944 and 2024.
M. pneumoniae is the primary cause of up to 40% of community-acquired pneumonia (CAP) cases in children [25]. M. pneumoniae epidemics occur every 3–7 years, however the major drivers responsible for the M. pneumoniae upsurges and the factors that influence transmission dynamics are still not understood [6]. A previous study reported a shift in the p1 subtypes during epidemic outbreaks of M. pneumoniae and suggested that a shift in the p1-type 1 and p1-type 2 subtypes may be a plausible driver of these M. pneumoniae outbreaks [26]. The delayed re-emergence of the M. pneumoniae epidemic was observed globally in 2024 [9], while the appearance of macrolide resistance, especially in China, during this period is a significant concern. Using metagenomic analysis, Li et al., 2024 identified 179 M. pneumoniae samples from East China, which were grouped into two epidemic clones: p1-type 1 (clade EC1) and p1-type 2 (clade EC2) [27]. While the EC1 (p1-type 1) clone is responsible for the elevated frequencies of macrolide-resistant M. pneumoniae (MRMP), the emerging EC2 (p1-type 2) clade now exhibits 100% macrolide resistance in East China [27].
Macrolides are the first line of treatment for infections caused by M. pneumoniae in many countries including the UK, and vaccine development, although still underway, has substantial hurdles. Hence, studying the presence of p1 subtypes and macrolide resistance in M. pneumoniae is crucial to further our understanding of seasonal epidemics.
Currently in the United Kingdom, there is no single surveillance system that fully captures national changes in M. pneumoniae activity. UKHSA has a multipartite and comprehensive respiratory surveillance program incorporating a wide range of community, syndromic and laboratory surveillance systems for monitoring known seasonal pathogens each winter [28]. An increase in the number of emergency department visits of patients with a diagnosis of pneumonia, especially those with M. pneumoniae infections was observed in England in the winter of 2023/2024 [28]. UKHSA’s voluntary surveillance database Second Generation Surveillance System (SGSS) is used to compile and present data related to M. pneumoniae laboratory detection. As infections caused by M. pneumoniae are not notifiable, this affects the surveillance data. Furthermore, only positive results are reported voluntarily, which may lead an underestimate of the number of cases or occurrence of disease caused by M. pneumoniae. Moreover, this is complicated by the fact that asymptomatic carriage, particularly in children can be missed during diagnosis.
In the UK, the methods for laboratory detection are based on serological detection of IgM or IgG levels in serum or detection of M. pneumoniae nucleic acid using nucleic acid amplification testing methods like PCR from clinical samples like throat swabs, nose or nasal swabs, bronchial aspirates, bronchoalveolar lavage, alveolar, nasopharyngeal aspirate, endotracheal aspirate, trachea, or sputum. However, these primary methods of laboratory detection do not report the presence of p1 subtypes, STs and macrolide resistance. A comprehensive surveillance typing method that can provide information on the p1 type, and the sequence types (STs) of the M. pneumoniae isolated during the epidemics is essential. This helps us understand the various p1 types that are circulating in the population and helps us gain a greater understanding of the STs circulating globally. Whole genome sequencing can help provide information on p1 typing, MLST profile, and can also help us understand the diversity in the p1 region.
It was observed that ST3 accounts for 43% (n = 141/328) of the M. pneumoniae strains present in the global data. This ST has been identified in China, France, Germany, UK, Korea, Taiwan, Japan, and the USA. The incidence of MRMP infections has increased worldwide, particularly in the Western Pacific region [29]. In Taiwan, a study by Hung et al. reported a Substantial increase in macrolide resistance rates from 12 to 24% during 2011–2016 to 54–88% during 2017–2020 [30]. A Substantial increase in macrolide resistance rates was also observed in the UK. In January and February 2024, macrolide resistance was identified in 5.2% (16 out of 309) and 4.7% (13 out of 276) of the samples, respectively; throughout the entire 2019–2020 season, resistance was detected in less than 1% (1 out of 110) of the samples (UKHSA 2024 M. pneumoniae surveillance report) [31]. In South China, 88.10% (n = 148) of the isolates from 2021 to 2023 were MRMP. This study also identified a high level of MRMP; 152/328 (46%) globally. For example, in South China, 100% (101 of 101) of the ST3 isolates from 2021 to 2023 were MRMP [32]. In the present study, we observed that MRMP is the most common in ST3 strains and is prevalent in Taiwan, Korea, Japan, China and the UK. The second most predominant MRMP sequence type was ST17 which was prevalent among strains in Taiwan. Of the 38 UK isolates in this study, 6 (16%) were macrolide resistant. Clinicians should be aware of macrolide resistant M. pneumoniae infection, both on initial and acquired infection. Genomic analysis indicated clonal expansion of the Lineage was Likely to have occurred. Whether this was with strains circulating in the UK or via importation cannot be determined with the current dataset and without improved genomic and epidemiological Surveillance. The travel history of the cases in this study was not known. Nonetheless, the presence of a novel sequence type in 2024 supports the theorem of naive population driving epidemics.
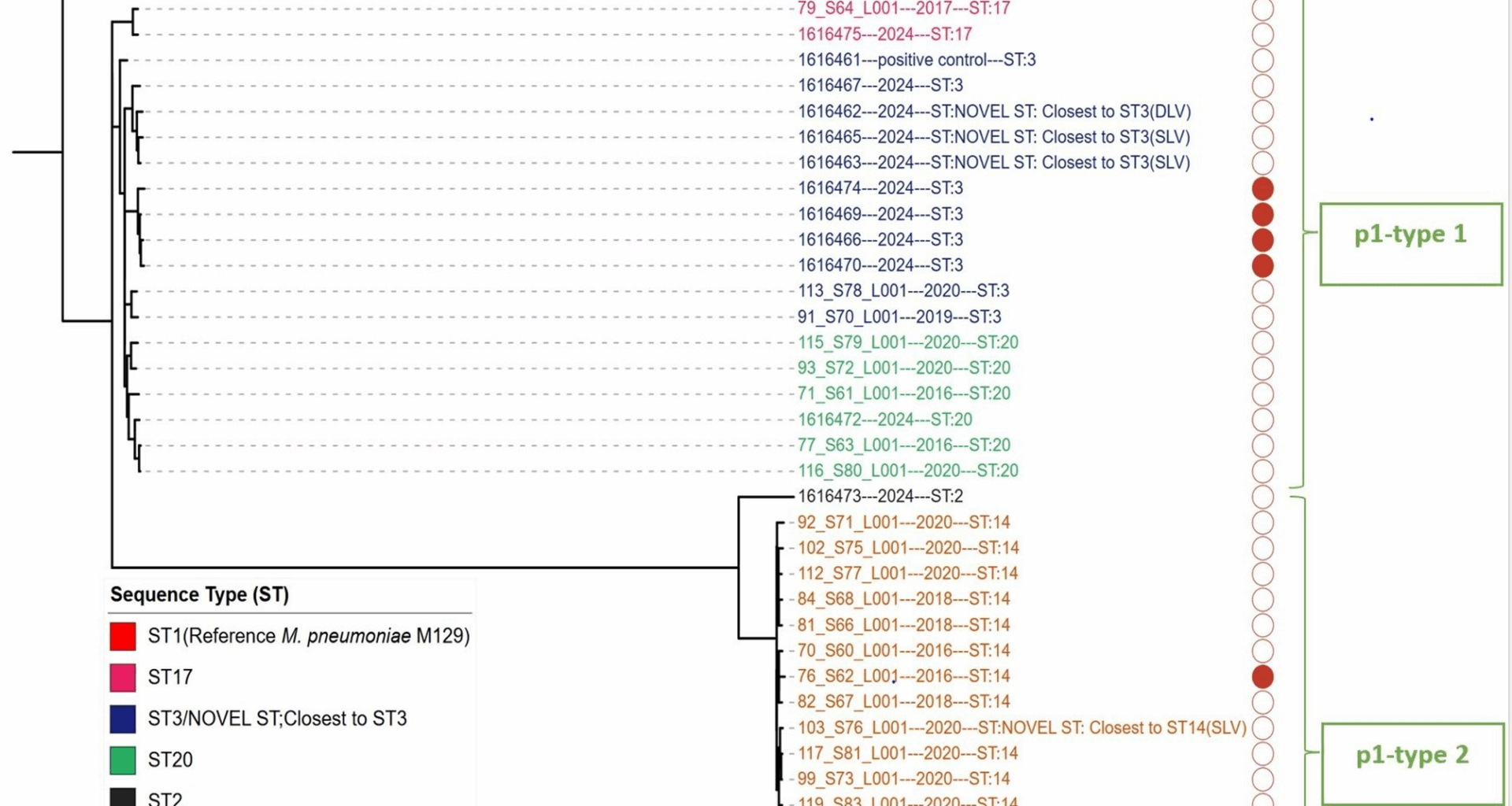
Core-genome phylogenetic analysis has been used to study genetic diversity in M. pneumoniae [16, 33, 34]. The two core-genome phylogenetic trees constructed and visualized in this study revealed notable findings (Fig. 1: Maximum likelihood phylogeny based on single nucleotide polymorphisms (SNPs) from 38 M. pneumoniae genomes, Fig. 5: Neighbour-joining phylogenetic tree based on a collection of M. pneumoniae isolates from UK and NCBI RefSeq database). First, the phylogenetic relatedness of the strains demonstrated a strong correlation according to p1 type. Second, each ST type was grouped by the same branch with some exceptions for the global data (Fig. 5: Neighbour-joining phylogenetic tree based on a collection of M. pneumoniae isolates from UK and NCBI RefSeq database). For example, within the global dataset, the ST3 strains were separated into two Subclades (Subclade 1 and Subclade 2). Subclade 1 (ST3) consists of the largest (99%) MRMP; including the most actively evolving strains in East Asia. Subclade 1 (ST3) was first detected in the UK in 2024 in this study, and the three ST3 MRMPs were clustered in Subclade 1 (Fig. 5: Neighbour-joining phylogenetic tree based on a collection of M. pneumoniae isolates from UK and NCBI RefSeq database and Fig. 6: Phylogenetic tree of Subclade 1 (ST3), constructed from a global collection of M. pneumoniae isolates). It was interesting to note the UK isolates situated within a clade of East Asian isolates in the recombination-corrected Subclade 1 phylogeny. This Suggests a successful international clonal expansion of this Lineage or multiple importation from other geographies. It is imperative to continuously monitor the spread and evolution of Subclade 1 in the UK, as well as in other countries, to monitor the further spread of this resistant lineage. WGS has proven to be a powerful tool in identifying this cluster, and its application should be expanded with global data from more isolates and countries to allow for further public health responses to threat antimicrobial resistance.
The present investigation identified the presence of CARDS TX in all M. pneumoniae isolates included in this study. The basic mechanism of CARDS TX is based on its property of cellular vacuolization and ADP-ribosyl transferase (ADPRT) activity. CARDS TX also causes cilia stagnation, nuclear fragmentation, and the release of inflammatory factors [35]. The presence of CARDS TX is related to the severity of disease [36] and it is significantly upregulated in humans during M. pneumoniae infection [37]. It is still unknown if variation in CARDS TX production within different M. pneumoniae strains affects disease severity [38]. We observed that all the p1-type 2 M. pneumoniae strains in the present study had a non-synonymous SNP (T1112G, I371S) which was absent in the p1-type 1 strains. (Supplementary Table 1: Patient and Genomic Data for 38 M. pneumoniae isolates from UK). A similar SNP was previously observed that differentiated the type 1 and type 2 strains [33]. It is not possible to elucidate if this SNP affected the activity of CARDS TX or affected the disease severity in the present study. But it is important to study this in future investigations to determine if variation in CARDS toxin in different strains and subtypes have any effect on strain virulence, persistence or severity in disease and clinical manifestations or if it may prove useful for immune-protection and vaccine design.
The high-molecular-weight proteins HMW1, HMW2, and HMW3 are essential for stability, adherence and gliding [39]. The internal structure of the surface adhesion complex of M. pneumoniae are made of terminal button that has HMW2, HMW3 and P65; bowl complex that has HMW2 and paired plates that is made of HMW1, HMW2, CpsG and HMW3 proteins [39]. In the present study it was observed that there was no mutation in HMW3 gene except for a unique C587T mutation in M. pneumoniae ST2 p1-type 2 strain. All the p1-type 2 M. pneumoniae ST 14 strains had two mutations G173A and T241C in HMW1 gene, while p1-type 2 M. pneumoniae ST 2 had only one mutation, T241C in HMW1 gene. All the p1-type 2 M. pneumoniae strains had a mutations T88C in HMW2 gene. There were no mutations seen in the HMW genes of the p1-type 1 M. pneumoniae strains. It is not possible to conclude if the mutation in HMW had any effect on the functioning or adhesion of M. pneumoniae p1-type 2 strains. Further research is warranted in this area to understand if mutations in these genes affects virulence adhesion or pathogenicity of different strains of M. pneumoniae.
Yu-Chia Hsieh et al., 2022 analysed 284 M. pneumoniae genomes for the investigation of recombination and found that the p1 adhesion gene is highly diverse and has multiple copies of the repeated regions RepMP4 and RepMP2/3 [34]. Different p1 variants/types are generated by DNA recombination between repetitive sequences (RepMP elements) in the M. pneumoniae genome [7]. Hence, we studied the putative recombination blocks present in M. pneumoniae isolates. This study detected putative recombination blocks covering an average of 2.3% of the genome, mainly in genes encoding for adhesin p1 family proteins and DUF16 domain-containing proteins (Supplementary Table 3: Sequence Coordinates of Putative Recombination Events). When performing core-genome SNP-based phylogenetic analysis, recombination regions were removed as recombination regions can have a variable effect on SNP analysis.
It is critical to recognize the limitations of this study. The sample size of the WGS study poses a significant drawback in terms of wider interpretation of the data. However, M. pneumoniae is difficult to culture. The organism can take up to 6 weeks to grow in broth culture and many clinical samples may test positive in the qPCR but are culture negative. Culture also depends on the type of sample, date of sample collection, type of transport medium used, and antibiotic treatment administered prior to sample collection. This difficulty in culturing isolates is also reflected in the relative paucity of publicly available M. pneumoniae genomes. Second, populations of M. pneumoniae can be found worldwide, but only genomes from 328 isolates were analysed in this study, which represented few countries and lacked global representation and geographical distribution.
The third limitation is that we did not study tetracycline-resistant M. pneumoniae (TRMP) or fluoroquinolone-resistant M. pneumoniae (FRMP). It is reasonable to assume that TRMPs and FRMPs may already be present in the genome, or that they are likely to evolve in the future, based on data from closely related Mycoplasma species. For example, fluoroquinolone-resistance is well documented in the closely related Mycoplasma genitalium, with dual resistance to macrolides and fluoroquinolones reported in greater than 50% of cases in parts of Asia [40]. Understanding the mechanisms of flouroquinolone resistance, and building a tool that could identify the likely point mutations related to TRMPs and FRMPs are vital for providing better guidance to inform clinicians of treatment options for MRMP isolates. To mitigate the challenges of Mycoplasma culture and reduce the turnaround time, culture-free NGS-based methods (e.g., NGS amplicon sequencing or target enrichment probe panels) or culture free metagenomics approaches could be used in the future for M. pneumoniae [41].