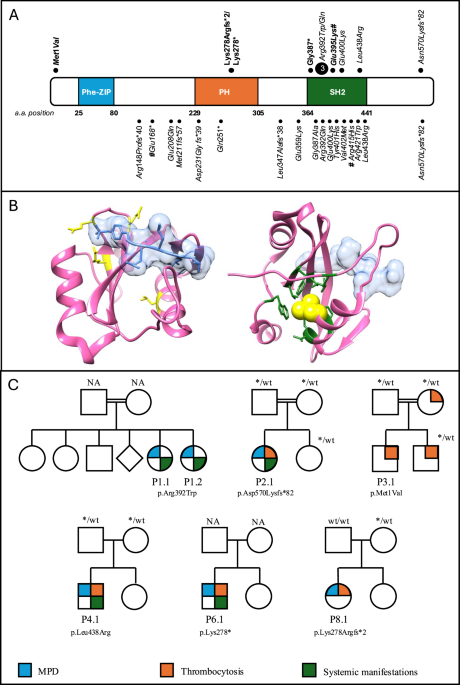
We here describe the clinical features of a cohort of 10 patients with germline SH2B3 variants. In our cohort, biallelic SH2B3 variants were associated in eight patients with MPD in the first weeks of life, including two with a monoallelic germline variant who acquired somatic LOH. Considering the age of onset of the phenotype, respectively at 0.2 and 0.7 years, the two patients most likely acquired LOH for the SH2B3 variant pre- or early postnatally and thus it is conceivable that the observed phenotype is related to the biallelic alteration of the gene. This suggest that the LOH occurred through a uniparental disomy (UPD) mechanism or a gene loss during early hematopoiesis, may lead to clonal expansion of hematopoietic cells. Interestingly, the VAF in blood for patient P8.1 who carries a monoallelic variant in germline material, was close to 100%, suggesting a near-complete replacement of normal hematopoiesis by the mutant clone. Unfortunately, detailed VAF dynamics pre- and post-treatment were not available for this patient. However, tracking changes in VAF over time could provide valuable insights into the clonal dynamics and the extent to which normal hematopoiesis can recover following the resolution of MPD. This observation is particularly novel and has significant implications for understanding the pathogenesis of SH2B3-related disorders. The mechanism of LOH is reminiscent of what is commonly observed in JMML, where somatic or acquired UPD is a well-recognized driver of clonal dominance. Future studies investigating the role of similar LOH mechanisms in SH2B3-related disorders may uncover parallels to other hematopoietic conditions and improve our understanding of disease progression and variability in clinical phenotype.
The clinical course is characterized by leukoerythroblastosis, monocytosis, low blast percentage in blood and marrow, splenomegaly, a normal karyotype and absence of somatic mutations. This presentation of the constitutional SH2B3-related disease is reminiscent of that observed in other MPD in neonates or infants, such as in patients with Down syndrome with somatic mutations in GATA1, or in RASopathies associated with germline mutations in CBL or PTPN11. In particular, MPD in CBL syndrome characterized by monoallelic germline mutation and LOH in hematopoietic cells, runs a self-resolving clinical course in the vast majority of cases. Interestingly, SH2B3 recruits the CBL protein via its conserved C-terminal tyrosine residue leading to an interaction between JAK-STAT and RAS-MAPK signaling. This interaction suggests that SH2B3 loss-of-function promotes myeloproliferation by activating the RAS-MAPK pathway through altered CBL modulation [2, 23, 24]. This was confirmed in transgenic mouse models, where the loss of both Lnk and Cbl leads to severe splenomegaly, extramedullary hematopoiesis, and exacerbated myeloproliferative characteristics [25] and reported in a previous case report [15]. Like in previous reports [9, 11, 12], the clinical course of neonatal MPD in patients with SH2B3 germline disease presented here was self-limiting in most cases. Three patients did not receive any therapy; two patients were treated with low dose cytoreductive therapy to ameliorate myeloproliferation. In two patients, chemotherapy and BCL-2 inhibition failed to control the disease, suggesting that some cases of SH2B3-related neonatal MPD may not run an indolent course. Considering the preclinical evidence of JAK-inhibition in SH2B3-mutant cells [10, 12], ruxolitinib could be an attractive therapy option. In the previous report ruxolitinib was effective in the resolution of splenomegaly and in the reduction of SH2B3 variant allele frequency [10]. In our cohort, ruxolitinib did not show efficacy in the treated patient. All patients in our cohort were alive at the last follow-up, including the three who received allogeneic HSCT, and none presented with abnormal blood counts except for thrombocytosis.
Indeed, the clinical presentation of these MPD also resembles JMML. Not surprisingly, SH2B3 variants have been identified in neonates suspected of JMML who lack a RAS pathway mutation, as reported in previous studies [9, 10]. The challenges and inconsistencies in categorizing these unique MPD in young children are further highlighted by the fact that MPD in CBL syndrome is traditionally classified as JMML. Arfeuille et al. reported on eight such patients from five families carrying biallelic germline variants [9]. Notably, while in our cohort the clinical phenotype shared features with JMML, the morphology, in particular the prominent atypical megakaryopoiesis and the absence of a significant increase in monocytic cell forms, was not characteristic of JMML [26]. However, since we evaluated BM specimens obtained in early infancy and not in the neonatal period, it is conceivable that the number of megakaryocytes in affected newborns is reduced as described by the French investigators [9], and the dysplastic features described arise later in early infancy.
In our cohort, following the resolution of MPD, patients with biallelic germline disease developed persistent thrombocytosis. One of these children underwent allogeneic HSCT for myelofibrosis at 10 years of age. In two other patients in this cohort (P 3.1, P 7.1), thrombocytosis diagnosed at 0.3 years and 4 years of age was the sole initial hematological presentation. Interestingly, the two children with monoallelic germline variants and LOH in hematopoietic cells had normal or moderately elevated platelet counts when neonatal MPD was diagnosed. The observation of thrombocytosis is consistent with the role of SH2B3 as a negative regulator of JAK/STAT signaling as previously demonstrated [4, 10, 12], also demonstrated by the increase in megakaryocytic progenitors, megakaryocytes, and platelets in hematopoietic tissue, along with an increase in erythroid progenitors, reported in Sh2b3-deficient mice [27]. Notably, the impact of SH2B3 LoF appears to be age-dependent with features of MPD in newborns and young infants and isolated thrombocytosis later in childhood, suggesting that SH2B3 variants have different effects on fetal and adult hematopoiesis. Remarkably, the mother of patient 3.1, who carries a heterozygous germline variant, has been known to have thrombocytosis since adolescence. Considering the clinical phenotypes associated with germline SH2B3 variants, it is puzzling that they can cause thrombocytosis or erythrocytosis in the absence of somatic mutations [28], while also cooperating with other driver mutations such as JAK2 or CALR to result in adult-type MPD [6, 7, 13, 14, 29], or PTPN11 to result in JMML. It must be said that one patient in out cohort presented a germline VUS in NF1; however, the patient did not develop any sign or symptoms of NF1-related conditions.
Given the lack of robust data on the long-term outcomes of this rare hematological disorder, a watch-and-wait strategy appears to be a reasonable initial approach, as suggested by others [9, 10]. In cases of extreme leukocytosis with pulmonary complications or significant organomegaly, cytoreductive therapy with 6-mercaptopurine or low-dose cytarabine may provide symptomatic relief. Later in the clinical course, disturbed megakaryopoiesis may lead to progressive myelofibrosis, resulting in an indication for HSCT.
Previous reports have described associations between germline SH2B3 variants and specific autoimmune conditions [9, 10, 17, 30]. In the presented cohort, six patients with biallelic SH2B3 germline variants and one of the two patients with a monoallelic germline variant and LOH in hematopoietic tissue displayed extra-hematopoietic symptoms, including IUGR, developmental delay, growth retardation, and dysmorphic facial features. Indeed, IUGR appears to be the most consistent constitutional phenotype. The underlying mechanism of this delay remains unclear, but one possibility is that it represents a hypoproliferative phenotype, potentially driven by an embryonic effect of SH2B3. To explore this, we examined available complete blood count data at birth in two patients who later developed MPD (P1.1 and P1.2), both of whom had normal hemoglobin levels. This very preliminary finding suggests that the phenotype may be more consistent with an intrinsic hypoproliferative mechanism rather than anemia-induced growth restriction. The presence of extra-hematological pathological features in both monoallelic and biallelic variant patients, suggest that a single hit may be sufficient to produce a phenotype. Indeed, the high incidence of these abnormalities in families with consanguineous parents may be a confounding factor, and larger cohorts will help define these clinical features [12]. Arfeuille et al. reported extra-hematological features as well, including cardiac involvement, which we did not confirm in ours [9]. Our report also underlines the frequent development of autoimmunity manifesting itself in childhood [9, 11, 12]. The role of SH2B3 in autoimmunity is suggested by earlier genome-wide studies in a variety of autoimmune disorders like rheumatoid arthritis, coeliac disease, hepatitis or diabetes type 1 [19, 31,32,33,34,35]. Moreover, it was shown that SH2B3 variants in patients with systemic lupus erythematosus are predominantly hypomorphic alleles failing to suppress interferon type II signaling via JAK2-STAT1 and impairing the negative selection of self-reactive B cells in mice [30]. This evidence may explain the high incidence of autoimmunity in these patients. However, further functional in vivo studies, including the exploration of biological markers related to cellular and humoral immunity, will be necessary to better characterize potential immune deregulations that contribute to their susceptibility to autoimmune diseases in late childhood. Apart from a distinct hematological phenotype, there are overlapping extra-hematopoietic features such as IUGR, growth retardation, and developmental delay. However, dysmorphic features appear more variable among individuals and larger cohorts will be essential clarify whether these findings define a consistent syndrome.
To summarize, germline SH2B3 LoF variants define a novel condition characterized by neonatal or in early infancy MPD which appears to be associated with extra-hematopoietic symptoms. Awareness of this genetic condition is key to a correct diagnosis and avoidance of unnecessary intensive therapy for the MPD. The establishment of large international registries spanning all age groups will be essential to better characterize the natural history of germline SH2B3 disease and to enhance our understanding of SH2B3 function across different ages and hematological manifestations.