Variants in NR6A1 cause an oculo-vertebral-renal (OVR) syndrome
We identified three rare NR6A1 variants in three families affected by uveal coloboma (COL005, COL034, COL171) with or without microphthalmia, cataract, and missing vertebrae through genome sequencing. In cases where multiple generations are affected, transmission is autosomal dominant with incomplete penetrance and variable expressivity (Fig. 1a). Clinical data for all the participants with a positive molecular result is shown in Table 1. No other candidate pathogenic variants in NR6A1 were identified in the NEI coloboma cohort consisting of a total of 224 probands (66 analyzed by genome sequencing, 57 by exome sequencing, and 101 by amplicon sequencing).
Fig. 1: Phenotypes associated with variants in NR6A1.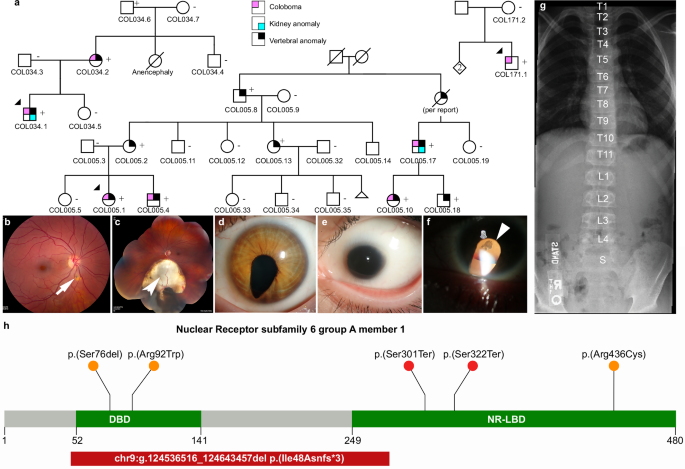
a Pedigrees of three families (COL005; COL034; COL171) from the NEI cohort demonstrating coloboma with or without microphthalmia and cataract, missing vertebrae, and congenital renal anomalies. Inheritance is autosomal dominant with incomplete penetrance and variable expressivity. b Linear pigmentary disturbance representing a forme fruste of coloboma (arrow) in COL005.1 (right eye). c Larger chorioretinal coloboma in the left eye of COL005.1 demonstrating a retinal tear in the far periphery (arrowhead). d Iris coloboma of the left eye of COL005.10. e Microphthalmia of the left eye in COL034.1. f Retro-illumination image of the left eye of COL171.1 demonstrating iris coloboma and posterior subcapsular cataract (open arrow). g Spine x-ray of COL005.4 demonstrating 11 thoracic (normal 12) and 4 lumbar (normal 5) vertebrae. h Schematic of NR6A1 variants detected in the NEI and UK Genomics England cohorts. + individual with variant, − individual without variant. DNA binding domain (DBD) and putative nuclear receptor ligand binding domain (NR-LBD) are noted (Q15406; InterPro).
Table 1 Phenotypic details of individuals carrying pathogenic variants in the NEI cohort
The proband of the family (COL005.1) presented at age 14, with bilateral uveal colobomas (Fig. 1b, c). Family history was notable for a younger brother (COL005.4), a second cousin (COL005.10), and a first cousin once removed (COL005.17) with uveal coloboma. The deletion breakpoints were in intron 2 and 6 removing the coding sequence for amino acids (aa) Ile48-Gly275 and likely causing a frameshift (p.Ile48Asnfs*3, Fig. 1h). The status of the heterozygous deletion was determined by breakpoint PCR among family members available, which revealed complete segregation with the missing vertebrae with an estimated LOD score of 3.6 (Fig. 1a, Supplementary Fig. 1). Four family members were also affected by coloboma in addition to missing vertebra, of which one (COL005.17) also had unilateral renal agenesis by report. The proband of the family (COL034.1) presented at age 11 months with bilateral uveal colobomas and microphthalmia OS (Fig. 1e). Prenatal history was remarkable for the inability of an ultrasound at 18 weeks to visualize the left kidney. Systemic testing demonstrated 10 thoracic vertebrae and left renal agenesis. Genome sequencing revealed a heterozygous c.274C>T p.(Arg92Trp) variant in NR6A1, which was found in the affected mother and unaffected grandfather (Fig. 1a, h). The proband of the family (COL171.1) presented at age 36 with bilateral colobomatous microphthalmia affecting the iris, retina/choroid, and optic nerve. Slit lamp exam was notable for bilateral microcornea, bilateral posterior subcapsular and nuclear cataracts, and missing zonules inferiorly OU (Fig. 1f). Genome sequencing revealed a heterozygous c.1306C>T p.(Arg436Cys) variant in the proband which was absent in his unaffected mother (Fig. 1h). Detailed study of the probands and their family members available for evaluation, were described in clinical vignettes in the Supplementary Notes. No convincing pathogenic variants in known coloboma genes were identified in any of these subjects.
Genome-first approach for NR6A1 variants corroborates microphthalmia, anophthalmia, coloboma (MAC) phenotypes
We performed an unbiased disease association analysis of rare pLoF variants using the UK100KGP dataset17. After removing variants resulting from calling artifacts or mis-annotation, only three pLoF variants were found in the cohort with approximately 126,700 alleles (Supplementary Data 1, Supplementary Fig. 2). We found three probands, Proband (A1) presented at age 30, with bilateral chorioretinal coloboma (forme fruste OD) and OS coloboma of the optic nerve (Supplementary Fig. 2b). Genome sequencing revealed a heterozygous c.965_980del p.(Ser322Ter), present in both the proband and her unaffected father. Proband (B1) presented at the age of 29, with a severe form of bilateral microphthalmia with a vestigial remnant of eyes, delayed motor development, intellectual disability, abnormal behavior, and schwannoma. This proband carried a heterozygous c.902G>A p.(Trp301Ter) variant. These two nonsense variants are expected to cause loss of protein function either through nonsense-mediated decay or truncation of the putative nuclear receptor ligand binding domain (NR-LBD, Fig. 1h). Proband D (Supplementary Data 1) had a disorder of sex development, carried variant c.288dup p.(Cys96TrpfsTer4), which was absent in either parent. His father was also affected with a disorder of sex development, suggesting that the NR6A1 variant is likely not associated with the condition.
The UK100KGP MAC cohort, which consists of 215 probands, was queried for rare missense and in-frame insertion/deletion variants. Proband C1, presented at the age of 25 with bilateral microcornea and coloboma affecting the iris, choroid/retina, and optic nerve. One brother had a similar condition by report. Both parents and the two other siblings of the proband had no history of coloboma by report. Genome sequencing revealed a heterozygous variant c.227_229del p.(Ser76del) present in the proband (Fig. 1h, Supplementary Data 1). This variant leads to an in-frame deletion of a serine within the Zn-finger motif. Within the three MAC patients we report, no candidate pathogenic variants were found in the known MAC genes present in the current Genomics England PanelApp (ocular coloboma v1.47, anophthalmia or microphthalmia v1.51, structural eye disease v3.79). Thus, these cases further support that rare variants in NR6A1 can cause MAC with reduced penetrance.
Molecular Modeling Suggests Missense Variants Disrupt Important Intramolecular Interactions
The NR6A1 amino acid sequence is well-conserved between human, mouse, and zebrafish; specifically, the residues Ser76, Arg92, and Arg436 are conserved across multiple species (Supplementary Fig. 3). To understand the effects, missense variants had on protein stability and function, we created an in-silico model of a complex of NR6A1 with DNA (Supplementary Fig. 4a). The AlphaFold model of NR6A1 is shown by the composition of Zn-finger (residues 60–172) and NR_LBD (residues 246–480) domains shown in orange and green, respectively. The rest of the model shown in gray is predicted as an irregular structure by AlphaFold. In wild-type (WT) NR6A1, a positively charged arginine residue 92 is predicted to interact with negatively charged DNA based on a zinc-finger protein model (Supplementary Fig. 4b). The R92W variant replaces the R92 residue with hydrophobic tryptophan (W), possibly interrupting the electrostatic interaction with DNA. The R436C variant affects the putative nuclear receptor ligand binding domain NR_LBD. In NR6A1, hydrogen atom 1HH2 of arginine R436 closely interacts with the oxygen atom of glutamic acid E388 (Supplementary Fig. 4c). The variant R436C breaks this bond and creates a cysteine residue which could form abnormal disulfide bridges in the variant protein, since residues C443, C391, and C422 are distanced at 8–12 Å from C436 in this variant domain as compared to 14–19 Å (C443–C391), 11.09 Å (C443–C422) and 4.52 Å (C91–C422) in the WT protein model.
Missense variants alter NR6A1 protein subcellular localization
To study the functional impact of the missense variants on protein localization in the cell, the R92W and R436C mutations were introduced in WT NR6A1 cDNA fused to a GFP coding sequence. All experiments were performed in context to the NR6A1 isoform NM_033334.4 and repeated at least three times. Transfection efficiencies were between 50-60% for the WT and variant constructs as analyzed by flow-cytometry and Western blotting (Supplementary Figs. 5, 6). The WT-NR6A1 when over-expressed in HEK293 cells was consistently observed to localize in the nucleus (Fig. 2a, b), consistent with a previous report22. The R92W variant, although nuclear, was not uniformly distributed across the nucleus. To study the localization of R92W variant puncta to the nucleolus, we performed immunofluorescence staining of NR6A1 wild-type and mutant isoforms with nucleolar marker FIBRILLARIN. As shown in supplementary Fig. 7, the punctae do not colocalize with FIBRILLARIN staining suggesting that the variant is not mis-localized to the nucleolus. In contrast, the R436C variant localized exclusively in the cytoplasm (Fig. 2a, b). The above-described localization pattern of the WT and variant isoforms was consistent in all transfected cells and across multiple rounds of transfection. Taken together these results suggest that both missense variants likely interfere with NR6A1 function due to improper subcellular localization.
Fig. 2: Subcellular localization of wild-type (WT) and mutant forms of NR6A1.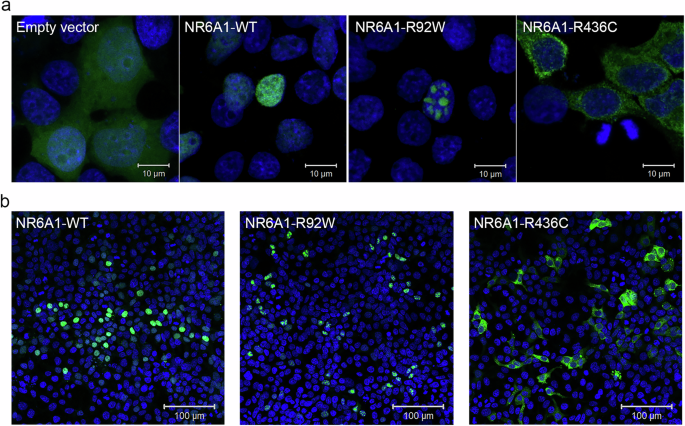
NR6A1 variant localization pattern was studied by overexpression in HEK293 cells and representative high magnification (63X) images are shown from three different trials (a) Scale bar = 10 µm. Low magnification images (b) scale bar 100 µm. The localization pattern for the WT and the two variant isoforms was observed to be consistent across three transfection experiments. (Cells counted: WT = 387, R92W = 350 and R436C = 217).
Expression pattern of mouse and zebrafish NR6A1 homologs suggests a role in early eye, kidney, and somite development
Analysis of bulk RNA-Seq datasets from ocular and non-ocular tissues demonstrates modest expression of NR6A1 in most tissues and relatively higher levels of expression in embryonic stem cells/induced pluripotent stem cells (compared to adult ocular tissues) and in bone marrow and testis systemically (Fig. 3a, d)23,24. Consistent with this observation, bulk RNA-Seq data from human fetal tissue shows that NR6A1 expression is highest in early stages of development, including the time of optic fissure closure in the first trimester25,26 (Fig. 3b). In the Human Retinal Cell Atlas single nucleus RNA-Seq dataset, NR6A1 is highly expressed in adult horizontal cells and low in microglia and RPE (Supplementary Fig. 8)27. Expression of NR6A1 is strongly correlated (>5 fold enrichment, p = 0.0024) with that of other coloboma-associated genes in fetal ocular tissues (Fig. 3c). This strength of enrichment was not seen in Genotype-Tissue Expression (GTEx) body tissue (p = 0.361) or adult eye tissue (p = 0.451)23,24. We note that several of the highly correlated genes-SALL4 (Duane-Radial Ray Syndrome), PAX2 (Papillorenal syndrome), ACGT1 (Baraitser-Winter Syndrome 2), SALL1 (Townes-Brocks Syndrome 1) can also present with congenital renal anomalies.
Fig. 3: NR6A1 gene expression in ocular and body tissues.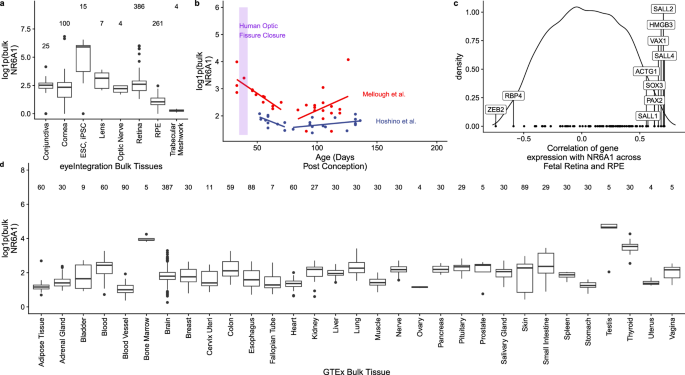
a Comparative levels of NR6A1 from publicly available bulk human tissue RNA-sequencing (RNA-Seq) datasets accessed on the eyeIntegration website (https://eyeintegration.nei.nih.gov/). On average, expression is higher in embryonic and induced pluripotent stem cells (ESC, iPSC, respectively) than in adult ocular tissues. b Bulk RNA-Seq data in human retinal fetal tissue from two studies suggests NR6A1 expression is highest in early stages of development, including the window of optic fissure closure (lavender box). NR6A1 expression is plotted against the tissue age (days post conception, dpc). A linear regression analysis was added for each paper’s data from the 40 to 80 dpc and 80 to 160 dpc samples. c The density correlation plot (closer to −1 and 1 is more negatively or positively correlated, respectively) shows ten notable coloboma associated genes with highly ranked correlations with NR6A1 expression across eyeIntegration curated fetal retina and RPE tissues. This enrichment was not seen in adult tissues. d Among systemic tissues, NR6A1 is expressed most highly in bone marrow and testes. The boxplots display the median, 25th and 75th percentiles, and 1.5 * interquartile range (IQR). Any data outside the 1.5 * IQR are plotted. In panels a and d the number of samples is given above the boxplots.
To establish plausible causation for NR6A1 variants, we studied the embryonic expression of its orthologs in mouse and zebrafish model systems at developmentally relevant time points. Previous work has demonstrated widespread expression of Nr6a1 in mouse at E8.5 and E9.5 (including the optic vesicle) that becomes nearly undetectable by E12.521. To study expression in the optic cup around the time of optic fissure closure, we used a probe that detects all validated transcripts of mouse Nr6a1 at embryonic day 10.5 (E10.5, early optic cup) and E11.5 (time of optic fissure closure). The manufacturer’s control probe against a bacterial sequence was used for reference (Supplementary Fig. 9). At E10.5, we noted diffuse low-level expression throughout the early optic cup and surrounding tissues that becomes significantly decreased by the time optic fissure closure commences (E11.5) (Supplementary Fig. 9). The level of expression in the optic cup is comparable to that observed in the brain vesicle, but higher than that observed in some areas of the surrounding eye mesenchyme.
In zebrafish, nr6a1 has two paralogs, nr6a1a and nr6a1b, both of which are maternally expressed28,29. At 11 hpf, when the optic vesicle evaginates, nr6a1a is widely expressed throughout the embryo, especially rostrally, showing less expression towards the posterior embryo axis (Fig. 4a, Supplementary Fig. 10a). Expression of nr6a1a is seen in heart and periocular tissue at 14 hpf (Supplementary Fig. 11); expression in the heart reduced by 16 hpf and is absent by 19 hpf (Fig. 4b, c). At 16 hpf, nr6a1a remains widely expressed, while becoming restricted to the ventral regions of the brain, epiphysis, periocular tissues, heart and in the notochord and neural tube (Fig. 4b); expression in the developing eye is reduced compared to the adjacent developing brain (Supplementary Fig. 10b). Notably, nr6a1a expression is absent from the neural-mesodermal progenitor region in the tail of zebrafish embryos, consistent with its role in the trunk differentiation program21. By 19 hpf the expression appears to decrease overall but remains present in the ventral brain regions, notochord, somites, and the pronephric duct (Fig. 4c). At 24 hpf, expression is prominent in the anterior diencephalon, tegmentum, midbrain, and along most of the length of the embryo in the neural tube; interestingly, expression is nearly absent from the neural retina and retina pigmented epithelia but is prominent in the lens (Fig. 4d–d”). After 26 hpf and up to 72 hpf we observed no detectable nr6a1a expression, consistent with published single-cell mRNA expression during zebrafish development28,29.
Fig. 4: Expression pattern of nr6a1a and nr6a1b paralogs in zebrafish.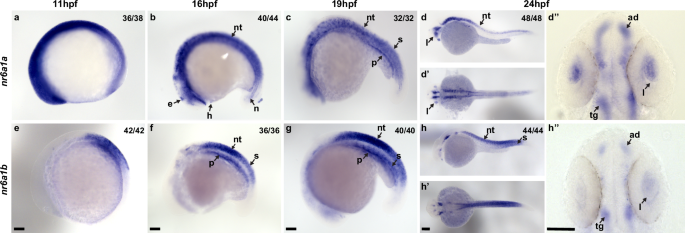
nr6a1a is expressed ubiquitously at 11 hours post-fertilization (hpf) (a). By 16–19 hpf (b, c) expression is present in multiple structures including the somites (S), neural tube (NT) and notochord (N). At 24 hpf, expression remains in the NT but is decreased in the S and N. Expression in the lens (L) is first noted at 19 hpf and is particularly prominent by 24 hpf (d–d”‘’). nr6a1b expression at 11 hpf (e) is in anterior trunk, localizing to neural tube and somites from 16 hpf (f) and 19 hpf (g). At 24hpf (h–h”) it remains expressed in the neural tube and somites, with faint expression can be seen in the lens. All embryos are oriented in a lateral view, anterior to the left and dorsal up, except (d’, d”, h’, and h”) shown in dorsal views. Scale bar = 100 µm. e-epiphysis, l-lens, p-pronephros, h- heart, n-notochord, s-somite, nt-neural tube, ad-anterior diencephalon, tg-tegmentum.
Unlike nr6a1a, nr6a1b expression at 11 hpf is limited to a patch in the posterior neuroectoderm of the embryo but excluded from the most caudal region (Fig. 4e). At 16 hpf and 19 hpf, nr6a1b expression is prominent in the neural tube, somites, and pronephric duct and, like nr6a1a, is excluded from the neural-mesodermal progenitor region in the tail (Fig. 4f, g). By 24 hpf, expression is decreased in most tissues but remains in the tegmentum, cranial ganglia, neural tube, and somites in the distal region of the trunk (Fig. 4h-h”). By 36 hpf and through 72 hpf, nr6a1b is notably expressed in the developing lens, brain, and cranial ganglions. (Supplementary Fig. 12). At 72 hp we also note faint expression in the retina and in the presumed RPE (Supplementary Fig. 12d, e).
Morpholino (MO) knockdown of zebrafish nr6a1a/nr6a1b recapitulates human phenotypes which are not rescued by pathogenic variant mRNA
All MO experiments were carried out following the guidelines set forth for their use in zebrafish30,31,32. These guidelines include: 1) use of two non-overlapping MOs (one translation blocking (TB), one splice blocking (SB)); 2) observation of a consistent phenotype with both TB and SB MOs for each paralog; 3) a correlation between dose of MO and phenotype, with lower concentrations of MO causing a milder phenotype; 4) validation of the efficacy of SB MOs by RT-PCR analysis; 5) lack of a phenotype with injection of a control MO; and; 6) partial rescue of the MO phenotype with co-injection of the corresponding human mRNA.
To test the functional consequences of nr6a1a and nr6a1b knockdown, we designed TB and SB MOs for each paralog of the gene. The sequence of the TB morpholinos does not overlap with the human mRNA sequence and is therefore unlikely to interfere with mRNA rescue experiments (Supplementary Fig. 13). Morphants were divided into four phenotypes: normal, mild (normal/near normal body axis with microphthalmia), moderate (slightly shortened and mildly curved body axis, microphthalmia ± coloboma and heart edema) or severe (significantly shortened and curved body axis, microphthalmia ± coloboma, heart edema) (Fig. 5a–d’, Supplementary Figs. 14, 15). Embryos were scored at 72 hpf (after optic fissure closure and initial stages of eye growth are normally completed) to ensure microphthalmia/coloboma represents a true phenotype and not because of developmental delay or undergoing growth compensation.
Fig. 5: Rescue of nr6a1+nr6a1b zebrafish morphant phenotypes with wild-type and mutant human NR6A1 mRNA.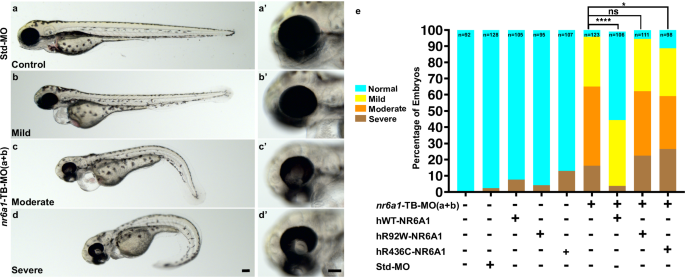
Controls (a, a’) have a straight body axis and the optic fissure (OF) is closed. The nr6a1+nr6a1b morphants that have a mild phenotype (b, b’) have close to a normal body with microphthalmia and heart edema; a moderate phenotype (c, c’) with a slightly bent body axis with smaller eyes, coloboma and a severe heart edema; and severe morphants (d, d’) have a curved body axis with smaller eyes, coloboma and heart edema. The morphant phenotype was rescued when the morpholinos were co-injected along with the human-NR6A1-wild-type mRNA. However, there was no significant rescue in the morphant phenotype when the morpholinos were injected with either R92W or R436C human disease-causing variants (e). Morpholinos were injected at 0.75 ng each (1.5 ng total). Embryos were imaged at 72 hpf. Scale bar = 100 µm. Statistical significance was calculated using Chi-square test and Fisher’s test. P value of nr6a1-TB-MO(a + b) V hWT-NR6A1 is nr6a1-TB-MO(a + b) V hR92W and hR436C are 0.0266 and 0.5169 respectively. Source data file has been provided for (e).
Knockdown of nr6a1a (Supplementary Fig. 14e) or nr6a1b (Supplementary Fig. 15e) with either TB-MO or SB-MO resulted in a significant number of moderate and severe phenotypes with few mild phenotypes. Although the effect of TB-MO and SB-MO were similarly potent for nr6a1b knockdown, the SB-MO had a stronger effect than the TB-MO for nr6a1a. SB-MO knockdown of the gene was validated for both paralogs by reverse transcription-PCR experiments (Supplementary Figs. 14f, 15f). MOs can elicit p53-activated cell death and cause non-specific defects33. We therefore tested MOs for both nr6a1 paralogues at all concentrations used with and without co-injection of a p53-MO. The phenotypic spectrum was not affected by co-injection of this p53 MO, suggesting widespread cell death was not the primary cause of our observations (Supplementary Fig. 16).
Overexpression of 100 pg of human NR6A1 mRNA in zebrafish shows no overt phenotype (Supplementary Fig. 17a, b). Co-injection of 2 ng and 1.25 ng of nr6a1a and nr6a1b, TB-MO respectively along with 100 pg of WT human mRNA (hWT-NR6A1), resulted in a rescue, with over 60% embryos exhibiting a normal/control-injected phenotype, thus validating our TB-MO’s (Supplementary Figs. 14g, 15g). In contrast, co-injection with either hR92W or the hR436C missense variants of NR6A1 identified in coloboma patients were significantly less effective in rescuing the zebrafish nr6a1a/b knockdown, indicating that the missense variants are deleterious (Supplementary Figs. 14g, 15g).
To study the effect of knocking down of both nr6a1a and nr6a1b zebrafish paralogues, we co-injected 0.75 ng of TB-MO for each paralog (1.5 ng total), resulting in a similar spectrum of eye and body axis phenotypes compared to the knockdown of individual paralogues (Fig. 5a–d, a’–d’). At 48 hpf, double morphants exhibited abnormal expression of nphs1/nephrin and nphs2/podocin, both of which are required for the formation, maturation and maintenance of kidneys34,35,36 (Fig. 6a–j). The spectrum of expression patterns included reduced, absent, midline and asymmetric expression of nphs1/2 (Fig. 6d, h) which persisted at 72 hpf (Supplementary Fig. 18).
Fig. 6: nr6a1(a + b) zebrafish morphants have kidney and vertebrae defects.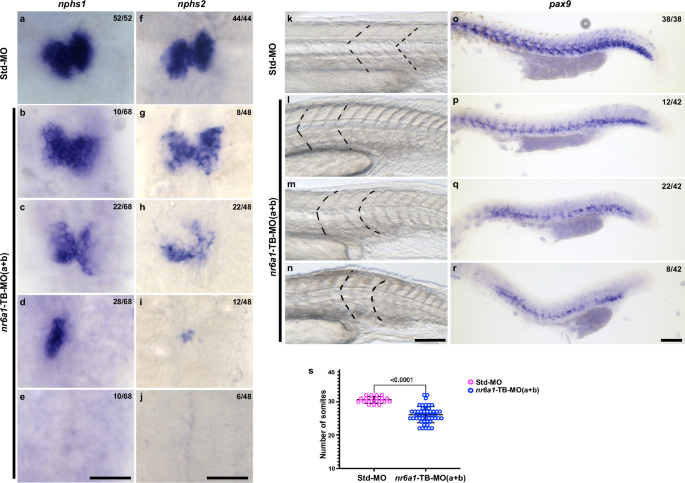
At 48 hpf, control embryos demonstrate bilateral expression of nphs1 (nephrin) and nphs2 (podocin) (a, f), markers of kidneys. Knockdown of nr6a1(a + b) resulted in a range of abnormal expression patterns including mildly reduced expression (b, g), moderately reduced expression (c, h), unilateral or midline expression (d, i) or absent expression (e, j) of nphs1 and nphs2. Somites (the early precursors of vertebrae) have a sharp, chevron shape with an approximately 90-degree angle in control zebrafish at 24 hpf (k). Knockdown of nr6a1a and nr6a1b resulted in a range of abnormal expression patterns including chevron shapes with obtuse angles (l), as well as mildly rounded (m) or flattened (n) somites. The sclerotome marker pax9 is expressed in a continuous angular pattern in the ventromedial portion of somites along the length of 24 hpf control embryos (o). Double morphant embryos exhibit a spectrum of abnormal patterns of pax9 expression including reduced levels of expression (p), patchy expression extending beyond the caudal tip of the yolk (q) and patchy expression ending near the caudal end of the yolk sac (r). nr6a1(a + b) morphants have reduced number of somites (s), statistical significance is calculated using two tailed t-test. Std-MO n = 20 and nr6a1-TB-MO(a + b) n = 50. Data is represented as difference between means of nr6a1-TB(a + b)–Std-MO ± SEM (−4.490 ± 0.5629). P value is s).
Vertebrae develop from the sclerotome of somites during development37,38. At 24 hpf, control embryos have chevron shaped somites, while morphants exhibit a spectrum of abnormal morphologies ranging from blunting of the chevron angle to more severe U-shaped or flattened somites (Fig. 6k-n). Double morphants also exhibit a significantly decreased number of somites, consistent with the missing vertebrae phenotype in several patients (Fig. 1, Fig. 6s). Similarly, the sclerotome marker pax939,40 is expressed in a uniform and regular pattern in the ventromedial region of somites of control embryos at 24 hpf (Fig. 6o). By comparison, double morphants show varying degrees of decreased and/or patchy pax9 expression (Fig. 6p-r). Overall, our results indicate that it is likely that nr6a1a + b morphant embryos have defective vertebrae development.
Injection of TB-MO’s, nr6a1(a + b), resulted in 16% and 49% embryos having severe and moderate phenotypes respectively; co-injection of 100 pg hWT-NR6A1 mRNA, resulted in >50% embryos having straight body and normal eye, however 19% of these embryos show, heart edema. Breaking down each phenotype separately, we rescued approximately 55% embryos for coloboma, 53% for body axis and 44% for heart edema. Neither the hR92W nor hR436C NR6A1 mRNAs resulted in significant rescue, of any of the described phenotypes, confirming the pathogenicity of these variants (Fig. 5e, Supplementary Data 5). Injection of 0.75 ng of either nr6a1a TB-MO or nr6a1b TB-MO resulted in a significantly milder phenotype, suggesting that co-injection of these had at least an additive phenotypic effect in the combined MO injection experiment (Supplementary Fig. 19).
A prior study reported that both overexpression and loss-of-function of nr6a1 can result in developmental phenotypes in Xenopus laevis41, we also evaluated the effect of injection of human NR6A1 mRNA on zebrafish development. Overexpression of 150 pg of hNR6A1 mRNA resulted in microphthalmia and heart edema with a straight body axis (n = 91/108) (Supplementary Fig. 17c, c’). At 200 pg, overexpression of hNR6A1 mRNA, phenotypes were more severe including colobomatous microphthalmia, heart edema and a bent body axis (n = 60/92), with 26% (n = 24/92) (Supplementary Fig. 17 d-f’), exhibiting noticeable shortening and loss of chevron-shaped somites (Supplementary Fig. 20); a minority of embryos (n = 8/92) developed no discernible eyes (Supplementary Fig. 17f, f’). Taken together, these experiments demonstrate that normal zebrafish eye development is sensitive to nr6a1 dosage and both reduced and increased nr6a1 expression result in developmental phenotypes analogous to human colobomatous microphthalmia.