Ethics statement
This study complies with all relevant ethical regulations and was conducted in accordance with the research permits and approvals granted by the respective institutions.
The INTERVENE Helsinki Biobank project was approved by the Helsinki University Central Hospital Ethics Board (HUS/230/2022 §69). The application meets the requirements for biobank research as defined by the Biobank Act (688/2012). The Bialystok Bariatric Surgery Study was approved by the Ethics Committee of the Medical University of Bialystok (approval number R-I-002/546/2015). This study was approved by the Institutional Review Board of the Icahn School of Medicine at Mount Sinai (23-00583). The Estonian Biobank (ESTBB) operates under the Human Genes Research Act, which has regulated its activities since 2000. Individual-level data analyses in ESTBB were conducted under ethical approvals 1.1-12/1409 and 1.1-12/2161 from the Estonian Committee on Bioethics and Human Research (Estonian Ministry of Social Affairs), using data according to release application 6-7/GI/18857 and 3-10/GI/11571 from the Estonian Biobank. The analysis in the Mass General Brigham Biobank (MGBB) was approved by the Mass General Brigham Institutional Review Board under protocol number 2018P001236. In Qatar Biobank all participants provided written informed consent for the biobank study and this study was approved by the Biobank Institutional Review Board number E-2024-QF-QBB-RES-QCC-00205-0280. UK Biobank data used in this study were obtained under approved application 78537.
For all other biobanks included in this study, additional ethical approval was not required as per their respective governance policies for secondary research using de-identified biobank data.
Study population
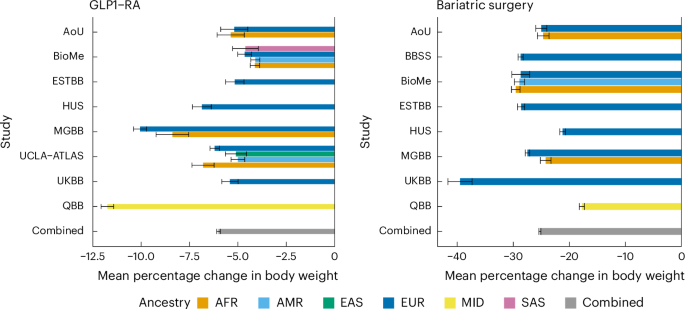
In the current study, we included samples from 10,960 individuals from the following nine biobanks: HUS (Finland; n = 633), ESTBB (Estonia; n = 464), UKBB (United Kingdom; n = 810), AoU (United States; n = 559), BioMe (United States; n = 2,170), MGBB (United States; n = 2,141), UCLA-ATLAS (United States; n = 1,445), QBB (Qatar; n = 2,383) and BBSS (Poland, N = 355). The biobank studies include samples from (hospital) biobanks, prospective epidemiological and disease-based cohorts. Follow-up covers a total of 20 years with the earliest study starting follow-up in 2004 (AoU) and the latest study ending follow-up in 2024 (BioMe). Below we provide a detailed description of the population selected and ancestry definitions in each study.
Inclusion and exclusion criteria for GLP1-RA analysis
For our GLP1-RA analysis we implemented the following inclusion and exclusion criteria. We required an initiation of a GLP1-RA treatment (ATC codes: A10BJ*, A10BX04, A10BX10, A10BX13, A10BX14) defined as the date of first medication purchase or medication prescription of GLP1-RA. Only first-time (GLP1-RA naive) users in the datasets were included. Individuals were required to be on a GLP1-RA treatment for at least 12 months. This was defined through prescription length, regular drug prescription refills or drug purchases, depending on the dataset and healthcare setting.
Individuals aged under 18 were excluded from the analysis. Furthermore, individuals were required to have at least one body weight measurement a maximum of 1 year before treatment initiation or within 14 days after initiation, if no measure before initiation was available. If we observed multiple weight measurements in this time frame, only the one closest to initiation was considered. This measurement defined our baseline weight variable. At least one body weight measurement between 26 and 52 weeks was necessary for each individual to be included in the analysis. Individuals were excluded if they underwent BS before or during the first year of GLP1-RA treatment.
Inclusion and exclusion criteria for BS analysis
For our BS analysis we implemented the following inclusion and exclusion criteria. Individuals were included if they underwent any type of BS. The respected code definitions for BS varied across countries and healthcare systems and can be found in Supplementary Table 22. The proportions of all procedures across cohorts can be found in Supplementary Table 23. Individuals aged under 18 years at baseline were excluded from the analysis. Analogously to the GLP1-RA analysis, individuals were required to have at least one body weight measurement a maximum of 1 year before surgery or within 14 days after it, if no measure before baseline was available. If we observed multiple weight measurements in this time frame, only the one closest to initiation was considered. This measurement defined our baseline weight variable. In addition, at least one body weight measurement between 26 and 208 weeks was necessary for each individual to be included in the analysis.
Outcome definitions
In our primary model, for both GLP1-RA and BS we defined the outcome to be the percentage change in body weight from baseline. The baseline body weight measured closest to T0 and between −52 weeks and +2 weeks from T0 was defined as W0. The second body weight (mW1) was defined as the median of body weight measurements between 26 and 52 weeks from T0 for the GLP1-RA analysis and between 180 and 1,460 days for the BS analysis. This was to enhance robustness against outlier measurements occurring in these intervals.
Therefore, the percentage change in body weight from baseline was calculated using
$$\% {\mathrm{Weight}}\; {\mathrm{change}}=\frac{m{W}_{1}\,-\,{W}_{0}}{{W}_{0}}\times 100.$$
In our secondary model, we used mW1 as an outcome.
Genetic exposures
We identified 15 genetic variants (SNPs) and two PGS of interest for our analysis. Among the included SNPs, three are functionally characterized GLP1R variants (rs10305492, rs146868158, rs6923761) associated with random glucose levels that have shown a decreased or increased response to different endogenous and exogenous GLP1-RA45. Seven SNPs are GLP1R missense variants (rs1042044, rs10305420, rs3765467, rs10305421, rs2295006, rs10305510, rs201672448) with a frequency >1% in at least one ancestry group in gnomAD (v.4.1)44. One SNP is the only robustly replicated lead genome-wide significant variant (rs429358, ApoE) in a genome-wide association study of BMI change over time41,61. Two SNPs are PCSK1 missense variants (rs6232, rs6235) that have been found associated with BMI variations46. And two SNPs (rs728996, rs17702901) were associated with an effect on excess BMI loss among BS patients36. A list of all genetic exposures and their frequencies across all major continental ancestry groups can be found in Supplementary Table 4. The included PGS are for BMI and T2D62. The PGS weights were taken from ref. 62 and obtained from UK Biobank. The scores were computed in each study using PLINK 2.0 (ref. 63). Because the PGS scores were derived from UK Biobank, we could not use the same score in UK Biobank, instead we used ref. 64.
Statistical model
We used a linear regression model to investigate the effect of the chosen genetic exposures on weight change after initiation of GLP1-RA treatment and BS, respectively. In the primary model we adjusted for the baseline weight, sex, age at initiation of treatment or surgery in years, the first 20 genetic principal components (PCs) and study-specific covariates, such as the genotyping batch. In the GLP1-RA analysis we additionally adjusted for the medication type in the drug class.
We defined % weight change as the outcome variable in the primary model.
Primary model:
$$\begin{array}{l}\% {\mathrm{Weight}}\; {\mathrm{change}}={\beta }_{0}+{\beta }_{1}\times {\mathrm{Genetic}}\; {\mathrm{exposure}}+{\beta }_{2}\times {W}_{0}+{\beta }_{3}\times {\mathrm{Sex}}\\\qquad\qquad\qquad\qquad+{\beta }_{4}\times {\mathrm{Age}}\; {\mathrm{at}}\; {\mathrm{initiation}}+{\beta }_5 \times {\mathrm{Medication}}\;(\mathrm{only}\; \mathrm{for}\; \mathrm{GLP}1)\\\qquad\qquad\qquad\qquad+\mathop{\sum }\limits_{k\,=\,6}^{25}{\beta }_{k}\times {PC}1:20\,+\mathop{\sum }\limits_{i=26}^{n}{\beta }_{i}\times {\mathrm{Study}}\;{\mathrm{specific}}\; {\mathrm{covariates}}\end{array}$$
Meta-analyses
We conducted ancestry-specific and multiancestry fixed-effect meta-analyses for each of the genetic exposures. Effect sizes were combined using fixed-effect inverse-variance weighting, as implemented in the R package meta65. For each exposure we reported the unadjusted P values, but considered as threshold for statistical significance P P \(\frac{0.05}{13}\), Bonferroni correction for 13 exposures tested) and P P \(\frac{0.05}{5}\), Bonferroni correction for 5 exposures tested). We tested for heterogeneity in effect sizes between ancestries using Cochran’s Q-test66 and by inspecting the I2 statistics67, which is defined as:
$${I}^{2}=\left(\frac{Q-\mathrm{d.f.}}{Q}\right)\times 100 \%$$
where d.f. is equal to the number of studies − 1.
Power calculation
To calculate the statistical power for the PGS effect sizes, we used a t-test for linear regression coefficient as implemented in the R package pwrs68 assuming a standard deviation for the outcome variable s.d.y = 7.8 (weighted combine s.d. across studies), a sample size of n = 6,750, a total number of predictors k = 25 and an adjusted r2 = 0.05 (coefficient of determination), to calculate power at P
For the coefficients of association with single variants, we estimated statistical power via the noncentrality parameter of the chi-squared distribution. We defined the noncentrality parameter as 2f (1 − f) n β2 where f is MAF, n is the effective sample size and β is the expected effect size69. We set a beta value of 0.3 (percent weight change from baseline) as the expected genetic effect on GLP1 response, based on findings from a recent large-scale study on genetic influences on drug response52.
Cohort descriptions and ancestry definitionsAll of Us
The AoU Research Program is a longitudinal cohort study aiming to enroll at least one million individuals across a diverse population in the United States. More than 245,000 individuals have genotype data. The sensitivity for single nucleotide variants was >98.7% and the precision was >99.9%.
Ancestry categories were based on gnomAD and inferred using principal components analysis (PCA) data. The patterns of ancestry and admixture were compared with self-identified race and ethnicity, and continuous ancestry inference using genome-wide genotypes resulted in concordant estimates. We restricted participants to an unrelated subset. The participants were further filtered based on the criteria described in the analysis plan (for example, \(\frac{\mathrm{Per}\; \mathrm{ancestry}\; \mathrm{minimum}\; \mathrm{allele}\; \mathrm{count}}{\mathrm{Sample}\; \mathrm{size}}\) after filtering on availability of weight measurements and prescription and/or BS).
Bialystok Bariatric Surgery Study
The BBSS is a prospective cohort study of patients undergoing BS at the First Clinical Department of General and Endocrine Surgery at the Medical University of Bialystok. This is the primary receiving center for patients referred for BS in the province of Podlaskie Voivodeship and the largest center by number of bariatric surgeries performed in northeastern Poland. This center specializes in several bariatric surgical techniques including RYGB, gastric banding and SG. For this study, we selected only patients who underwent SG because it represents the vast majority (>90%) of all interventions performed at the center and to eliminate confounding variation in surgical technique. The BBSS began in 2015 and consisted of a battery of baseline tests established one month before the intervention and repeated at 1-, 3-, 6- and 12-month follow-up clinical visits. Subsequently, patients are examined every year after the first year. At each visit, all subjects underwent physical examination, body composition analysis and blood testing, as well as completed diet and physical activity questionnaires. All subjects give their informed consent for inclusion before participating in the study. The study is conducted in accordance with the Declaration of Helsinki, and the protocol was approved by the Ethics Committee of the Medical University of Bialystok (Project identification code: R-I-002/546/2015).
Genetic ancestry in the BBSS cohort was determined using a combination of self-reported race or ethnicity and genotype-based analysis. Self-reported race or ethnicity information was collected during baseline assessments. Additionally, genetic ancestry was further refined through genotype clustering analyses using reference populations from the 1000 Genomes Project.
BioMe
The BioMe Biobank, founded in 2007, is an ongoing EHR-linked biorepository that enrolls participants nonselectively from across the Mountain Sinai Health System. More than 60,000 participants have been recruited from >26 outpatients sites located in Manhattan and Queens, and recruitment is ongoing. We restricted participants to an unrelated subset (based on second-degree or greater KING-derived kinship coefficients) who had been genotyped with either the Global Screening Array or Global Diversity Array. We then filtered this population to those adhering to the criteria described in the analysis plan (for example, \(\frac{\mathrm{Per}\;\mathrm{ancestry}\;\mathrm{minimum}\;\mathrm{allele}\;\mathrm{count}}{\mathrm{Sample}\;\mathrm{size}}\) after filtering on availability of weight measurements and prescription and/or BS).
Genetically determined ancestry group based on ADMIXTURE with K = 10 (where K refers to the number of ancestral clusters inferred in the ADMIXTURE analysis), with grouping based on which 1000 Genomes phase 3 reference samples (with superpopulation labels as reference) showed the highest proportion for a given class. The choice for K = 10 was based on a combination of minimizing cross-validation errors for admixture analysis, as well as minimizing the Frobenius distance of each ADMIXTURE matrix K with the matrix outputted by Genetic Relationship and Fingerprinting.
Estonian Biobank
ESTBB is a population-based biobank that involves more than 212,000 adult participants (about 20% of the Estonian adult population). The Estonian Biobank Project was initiated in 1999; data collection began in 2002. Initially, participants were recruited by general practitioners across Estonia from among individuals visiting general practitioners’ offices and hospitals. Consenting adults donated blood samples for DNA extraction, underwent anthropometric, blood pressure and resting heart rate measurement, and provided extensive information on demographic characteristics, genealogy, education, occupation and lifestyle, as well as health status and medical history70. From 2017 onward, the process of joining ESTBB was simplified: new joiners were required to sign an online consent form, visit a healthcare provider or pharmacy to provide a blood sample, and fill in an online questionnaire. Additional data are collected through linkage with various national databases and registries and through subsequent studies involving different subsets of participants.
Participants were genotyped using the Illumina Global Screening Array v.1.0, v.2.0 and v.2.0_EST. Samples were genotyped and PLINK format files created using Illumina GenomeStudio v.2.0.4. Individuals with call rates below 95% or whose sex in phenotype data did not match X chromosome heterozygosity were excluded. Before imputation, variants were filtered by call rate P −4 (autosomal variants only) and MAF 71.
We aligned participants to 21 major ancestry groups similarly to Privé72. We removed participants with nonEuropean assigned group ancestry, keeping Europeans, Finns and Italians.
Helsinki University Hospital/Helsinki Biobank
The Helsingin Biopankki (Helsinki Biobank) was founded in 2015 by HUS Helsinki University Hospital, University of Helsinki, the Joint Municipality for Health and Social Services in the Kymenlaakso Valley (Kymsote) and the South Karelia Social and Health Care District (Exote), and is the largest hospital biobank in Finland. Approximately 80,000 patients have given a blood sample based on biobank consent and used for research purposes. Diagnostic formalin-fixed paraffin-embedded tissue samples from approximately one million patients have been transferred to the biobank’s sample collection. The biobank’s samples were combined with clinical patient data from the hospital’s database (HUS Datapool) in accordance with the description in the research plan recommended by the ethics committee. Data that are run together can usually include the sampling date, the donor’s gender, histological and cytological examinations, diagnoses, care measures and laboratory examination results.
Mass General Brigham Biobank
Mass General Brigham is an integrated healthcare system located in Boston, Massachusetts for more than 1.5 million individuals per year. The MGBB has recruited more than 140,000 participants since 2008. More than 65,000 participants have genotype data sequenced using the Illumina Multi-Ethnic Genotyping assay or the Illumina Global Screening Array. All genotyped biospecimens had at least 99% call rate per array and were confirmed for sex concordance using sex in EHR and sex computed from array data.
Ancestry was defined using high-dimensional PCs, specifically top 30 genetic PCs and a network-based clustering approach. We restricted participants to an unrelated subset. The participants were further filtered based on the criteria described in the analysis plan (for example, \(\frac{\mathrm{Per}\;\mathrm{ancestry}\;\mathrm{minimum}\;\mathrm{allele}\;\mathrm{count}}{\mathrm{Sample}\;\mathrm{size}}\) after filtering on availability of weight measurements and prescription and/or BS).
UCLA-ATLAS (Atlas Biobank)
ATLAS is an EHR-linked biobank across multiple institutions in the UCLA Health System. After participants signed informed consent for participation to the UCLA-ATLAS Community Health Initiative, biological samples in the form of de-identified blood samples were collected and subsequently processed for DNA extraction and genotyping. The data analyzed in this manuscript draw from a ‘frozen snapshot’ of ATLAS data encompassing all available samples up to 15 February 2024, totaling n = 53,829. The corresponding EHR data for ATLAS participants were sourced from the UCLA Data Discovery Repository. This repository operates under the auspices of the UCLA Health Office of Health Informatics Analytics and the UCLA Institute of Precision Health. Patient Recruitment and Sample Collection for Precision Health Activities at UCLA is an approved study by the UCLA Institutional Review Board. Extensive details on ATLAS genotyping and quality control were previously reported in ref. 73. In short, participants in the ATLAS initiative were genotyped using a custom genotyping array constructed from the Global Screening Array with the Infinium Global Screening Array-24 Kit (Illumina), a multidisease drop-in panel aligned with the GRCh38 assembly. SNPs were subjected to removal if they were unmapped, strand-ambiguous, duplicates or exhibited >5% missingness. Samples with missingness exceeding >5% were excluded, as were duplicates (including identical twins or triplets) with preference given to individuals with the lowest missing rate in each pair. Imputation to the TOPMedFreeze5 was conducted using the Michigan Imputation Server74. After imputation, SNPs with a quality score (R2) below 0.90 were excluded, for PGS calculation SNPs with a MAF below 0.1% were filtered out.
The genetic ancestry of ATLAS individuals was estimated by assessing their proximity to 1000 Genome super populations on the PC space. We computed the top 20 PCs using bigsnpr R software with default parameters. Subsequently, leveraging the superpopulation label and PCs of the 1000 Genome individuals, we train a K-nearest neighbors model to assign genetic ancestry labels to each ATLAS individual. To optimize model performance, we used tenfold cross-validation to select the optimal hyperparameter (k) from a range of values (k = 5, 10, 15, 20). In instances in which an individual was assigned to multiple ancestries with probability >0.5 or was not assigned to any cluster, their ancestry was labeled as unknown. We clustered 94% of the ATLAS participants into one of the five 1000 Genome superpopulation (African (AFR), admixed American (AMR), East Asian (EAS), European (EUR), Middle Eastern (MID), South Asian (SAS)) while the remaining individuals’ ancestry could not be ascertained.
UK Biobank
The UKBB is a large-scale biomedical database and research resource containing in-depth genetic and health information from half a million UK participants. Upon signing informed consent, participants provided biological samples, including de-identified blood samples, which were processed for DNA extraction and genotyping. The dataset analyzed in this manuscript is derived from a ‘frozen snapshot’ of the UKBB data, encompassing all available samples up to 31 January 2024, totaling n = 502,536. Corresponding EHR data for UKBB participants were sourced from the UK Biobank Resource, which operates under the auspices of the UK Biobank Steering Committee and has received ethical approval from the North West Multi-centre Research Ethics Committee. Detailed information on UKBB genotyping and quality control has been previously reported75. Samples were genotyped at the Affymetrix Research Services Laboratory in Santa Clara, California, USA. Upon receipt of a 96-well plate containing 94 UK Biobank samples, Affymetrix added two control individuals (from 1000 Genomes) to the same well positions on each plate. Genotypes were then called from the resulting intensities in batches of ~4,700 samples (~4,800 including the controls) using Affymetrix Power Tools software and Affymetrix Best Practices Workflow. After genotype calling, Affymetrix performed quality control in each batch separately, to exclude SNPs with poor cluster properties. If an SNP did not meet the Affymetrix prescribed QC thresholds in a given batch, it was set to missing in all individuals from that batch. Affymetrix also checked sample quality (such as DNA concentration) and genotype calls were provided only for samples with sufficient DNA metrics.
The ancestry composition of UKBB participants was defined based on self-reported ethnicities and genetic PCA. Participants self-reported their ethnic backgrounds into categories such as White, Mixed, Asian or Asian British, Black or Black British, and Other ethnic groups, with the majority (~94%) identifying as White British. Genetic PCA was used to capture the underlying population structure and define genetic ancestry more precisely, categorizing participants into groups such as European, South Asian, African, East Asian and other ancestries. This approach ensured a nuanced understanding of the genetic and demographic diversity in the cohort, enabling robust and reliable biomedical research.
To capture population structure specific to the UKBB cohort, we performed principal component analysis of ~150,000 UKBB samples using ~100,000 SNPs. These PCs can be used to identify samples with similar ancestry or to control for population structure in association studies.
Qatar Biobank
This study was based on the second release of 14,060 Qatari participants (all from the same ancestry, MID) from the population-based Qatar Genome Program (QGP). Ethical approval was provided by the institutional review board of the QBB (Protocol no. QF-QBB-RES-ACC-00205). A detailed description of the recruitment process and collection of phenotypic data by the QGP and the QBB was previously reported76. A signed consent was obtained from all included participants by the QBB, and WGS data used in the current study were obtained from the QGP as previously described77. All samples were sequenced on Illumina HiSeq X instruments to obtain WGS with a target average depth of coverage of 30×. The read mapping, variant calling and joint variant calling were performed using Sentieon’s DNASeq pipeline v.201808.03, following the BWA-GATK Best Practice Workflow and using the GRCh38/hg38 reference genome. The produced gVCF were jointly called to produce one multisample VCF file for the whole cohort. Clinical data are extracted from the EMR of the Hamad Medical Corporation.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.