Previous large-scale GWAS studies have independently identified numerous genetic variants associated with the obesity-related traits such as BMI and WHR13,14 as well as IS indices15,16,17. In this study, we investigated the shared genetic factors between BMI/WHR and fasting and OGTT based IS indices and examined the impact of BMI adjustment on these associations. Our analysis showed that after adjusting for BMI, the number of BMI-associated variants significantly linked to IS indices, indicating that many genetic associations between BMI and IS are mediated through adiposity. The BMI and WHR variants were generally associated with both increased and decreased IS, with effects varying across fasting and OGTT-based IS indices. Additionally, WHR-associated variants exhibited sex-specific effects, with a stronger influence observed in females, particularly with postprandial IS indices following BMI adjustment.
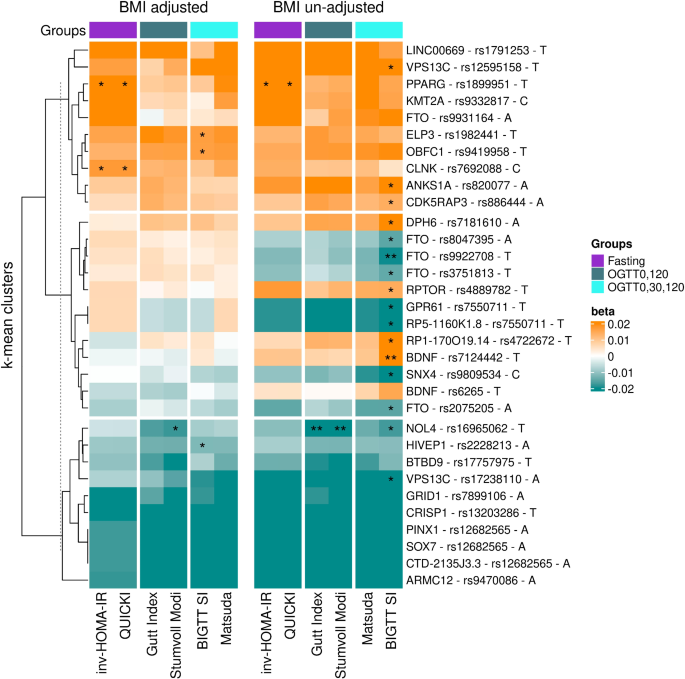
Previous research has established that a high BMI polygenic risk score is a risk factor for T2D18, which is primarily mediated through onset of prediabetes and insulin resistance (IR)19. Consistent with this, our study reveals a pattern where genetic variants linked to obesity-related traits tend to be associated with reduced IS in models unadjusted for BMI. Many IS-associated variants, including those in FTO, BDNF and other loci, showed significant associations prior to BMI adjustment. However, after adjustment, most associations were attenuated, except for variants in PPARG and NOL4, which retained their significance. This is in line with prior studies where genetic variants in the FTO locus lost their association with IS index (Stumvoll index) after BMI adjustment20. These results support the hypothesis that genetic influences on IS, particularly postprandial IS, are largely mediated through BMI-dependent mechanisms, reinforcing the shared genetic predisposition between adiposity and glucose metabolism, as supported by studies showing a strong link between BMI and IR21.
Our results further highlight that fasting-based IS indices may have a stronger genetic component than postprandial IS indices. Associations that remained significant after BMI adjustment were primarily observed with fasting indices, suggesting that genetic factors may exert a more direct influence on fasting IS, whereas postprandial IS is more susceptible to environmental and BMI-mediated effects. This is consistent with previous study demonstrating that fasting IS has strong genetic component, while postprandial IS is more influenced by other non-genetic factors9. Notably, some variants became significantly associated with IS indices only after BMI adjustment, emphasizing the complexity of IS regulation. These findings suggest that different IS indices capture distinct physiological mechanisms, with fasting IS being more directly influenced by genetic predisposition, while postprandial IS is more intertwined with adiposity and fat distribution. Given the well-established link between higher BMI and increased IR due to greater adipose tissue accumulation, these results underscore the interplay between genetic susceptibility, body composition, and IS.
The presence of IS-improving variants among BMI-associated loci confirms the existence of “metabolically healthy obesity” genetic variants. Previous studies have shown that some genetic variants linked to increased adiposity confer favourable cardiometabolic profiles, implying a genetic basis for metabolically healthy obesity. These findings support the idea that certain individuals, due to their genetic makeup, can store fat in a way that minimizes ectopic lipid accumulation in vital organs, preserving metabolic health. This aligns with previous evidence that adiposity-promoting variants can also have protective effects on metabolism, highlighting the importance of understanding genetic heterogeneity within obesity-related traits22.
Interestingly, variants associated with both increased and decreased IS were linked to diverse metabolic consequences, challenging the assumption that all BMI- and WHR-associated variants generally contribute to metabolic dysfunction. For instance, PPARG, a key gene in lipid and glucose metabolism23,24, was positively associated with fasting-based IS and lower T2D risk, supporting its role in beneficial metabolic outcomes. Similarly, VPS13C variants exhibited protective effects on IS and were linked to decreased T2D and stroke risk. Conversely, variants in FTO, VPS13C, and GPR61 loci were associated with reduced IS and increased T2D risk, consistent with expected metabolic dysfunction. Interestingly, variants rs17238110 and rs12595158 within the same locus VPS13C, displayed opposite effects on IS as well as T2D, CAD and stroke, illustrating the complexity of genetic influences within the same genomic region.
We also identified variants in AC022431.2 and BCL2L11 that were associated with decreased IS and an increased risk of T2D, CAD, and stroke, suggesting their potential role in glucose metabolism regulation. BCL2 has previously been linked to postprandial IS in BMI-adjusted models15. While the precise mechanism by which BCL2L11 influences IS remains unclear, the BCL2 family of proteins is known to regulate apoptosis by controlling mitochondrial permeability25. Similarly, AC022431.2, though not well-characterized, is highly expressed in adipose tissue, suggesting a possible role in metabolic function. Conversely, a variant in JAZF1, which was associated with increased IS, was linked to an elevated risk of T2D while showing no apparent effects on CAD or stroke. JAZF1 has previously been implicated in reduced insulin release and beta-cell dysfunction26. This paradoxical association of JAZF1 with both improved IS and higher T2D risk may be explained by a compensatory mechanism where reduced insulin release is offset by enhanced IS27, yet still contributes to increased T2D risk.
A variant in THADA was associated with increased IS but displayed a distinct risk profile for cardiometabolic diseases. THADA has been implicated in energy homeostasis, thermogenesis, pancreatic beta-cell function, and T2D28,29,30,31. Its dual role, enhancing IS while also increasing T2D risk, highlights its pleiotropic nature and diverse metabolic effects. The observed increase in IS could be explained by a compensatory mechanism, where improved insulin action offsets beta-cell dysfunction, potentially contributing to an increased risk of T2D. However, the inverse association with CAD remains unclear and warrants further investigation.
This study has some limitations. The sample size, while robust, may limit the statistical power of association analyses, and our findings have not been validated in diverse populations. Although the homogeneity of our sample minimizes ethnic confounders, it also restricts the generalizability of our results. Furthermore, adjusting for BMI in genetic analyses introduces complexities, as BMI itself is influenced by genetic variants and impacts IS, leading to potential collider bias32. This is particularly relevant for WHR-associated variants, given that they were identified in a BMI-adjusted framework in prior studies. While we acknowledge these potential biases, our results provide valuable insights into the genetic interplay between obesity and IS, warranting further validation and mechanistic exploration.
In summary, our findings demonstrate that genetic predisposition to higher BMI significantly impacts IS, with distinct effects on fasting and postprandial IS indices. Most BMI-linked variants were associated with IS in unadjusted models, but these associations largely diminished after BMI adjustment, suggesting their effects are mediated through adiposity. Conversely, WHR-associated variants, particularly in females, showed significant associations with OGTT-based indices after BMI adjustment, underscoring sex-specific genetic influences on IS. Notably, our results consolidate the notion that not all obesity-associated variants negatively impact metabolic health; some confer protective effects on IS, reinforcing the concept of metabolically healthy obesity. This study provides insights into the intricate genetic relationships between obesity and IS, offering a deeper understanding of the mechanisms driving metabolic health and disease. Understanding these relationships is crucial for the development of targeted interventions aimed at improving metabolic health through personalized genetic insights.