Studies performed using traditional GWAS and meta-GWAS approaches on large patient populations (6,450 cases and 53,764 cases) respectively identified a single locus and three loci associated with long COVID25,26, although there was no statistical replication of the findings between these studies.
The original combinatorial analysis of Sano’s GOLD cohort identified 9,068 genetic disease signatures and 73 genes that were significantly enriched in two small UK-based long COVID patient cohorts (Severe n_cases = 459 and Fatigue Dominant n_cases = 477)30. In this original analysis, 28/43 genes found in the Severe cohort were also significantly associated with disease in the Fatigue Dominant cohort, and 25/35 genes from the original Fatigue Dominant analysis were also associated in the Severe cohort. 25 genes (15 from Severe and 10 from Fatigue Dominant) were found to be unique to those cohorts.
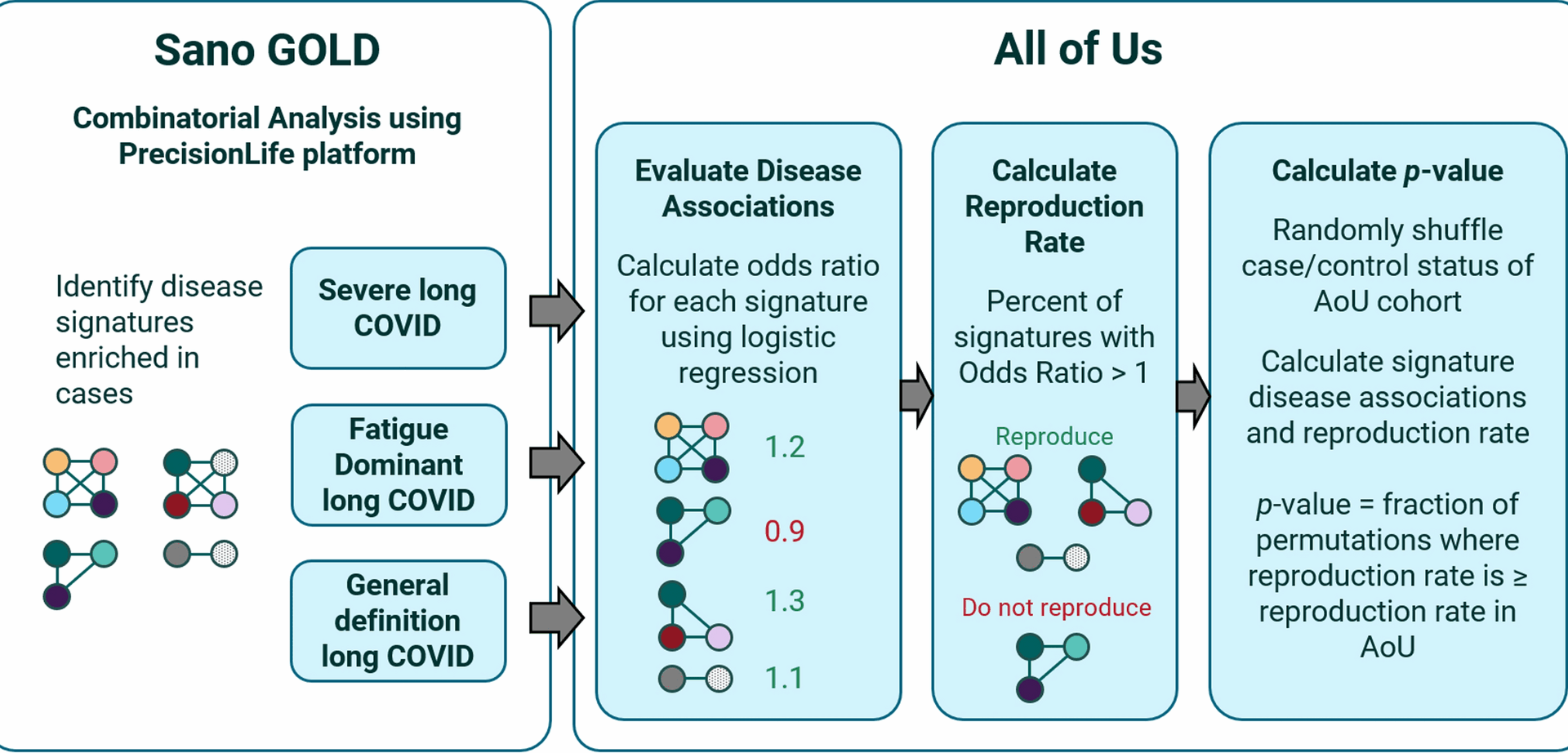
92% of the genes and 60–83% of the medium/high-frequency disease signatures from the Sano GOLD results that are also represented in the AoU dataset were positively correlated with long COVID in this independent US-based population. For disease signatures that occur in at least 5% of patients, between 77 and 83% were positively correlated with long COVID prevalence in both the Sano GOLD and AoU cohorts, far more than we would expect to randomly observe if the signatures were uncorrelated with disease biology. Although we defined a ‘reproducing’ signature as one that has any odds ratio greater than 1, most reproducing signatures have relatively large odds ratios in AoU, indicating a strong association with increased disease prevalence.
At least five of the disease signatures found in Sano GOLD were individually significantly associated with increased prevalence of long COVID in the AoU population. The significant enrichment of positively-associated disease signatures further confirms that many additional signatures are non-randomly associated with disease but cannot be individually validated due to the very low statistical power provided by the small number of long COVID patients in the dataset (n = 413). Together these results demonstrate a significant enrichment and reproduction of disease signal, broadly validating the results of the original analysis.
Replication of individual loci was not shown by the previous, much larger GWAS studies. The fact that five disease signatures did directly replicate, even with such a small sample size, suggests that if we were able to analyze cohorts closer to the size of the GWAS studies, it is likely that many more signatures would be directly replicated. As the All of Us long COVID cohort increases in size we will revisit this analysis, which will allow us to estimate the sample size needed for this broader replication.
Importantly, the results provide strong supporting evidence for a much broader range of genetic associations with long COVID than has been uncovered by GWAS studies to date. This provides evidence highly consistent with a strong biological basis of the disease and the hypothesis that patients’ genetics influence their susceptibility to developing long COVID (and their predominant symptoms) following recovery from acute SARS-CoV-2 infection.
The AoU ancestry distribution differs significantly from the mainly (> 91%) white British patient cohort used in the original combinatorial analysis. Disease signature reproducibility rates are very strong in the sub-cohort of self-identified white patients, as expected given the similarity in ancestry between that cohort with the original Sano GOLD dataset. Signature reproducibility rates are lower in sub-cohorts of self-identified black/African-Americans and Hispanic/Latinos, but we still observe significant enrichment of disease signatures despite very small sample sizes.
This therefore represents the first reproduction of long COVID genetic associations across multiple populations with substantially different ancestry distributions. Given the degree of reproducibility of results across diverse populations, these findings may have broad clinical application which could promote better health equity.
The lower signature reproducibility rates among the self-identified black/African American and Hispanic/Latino sub-cohorts relative to the self-identified white sub-cohort could be caused by a combination of the lower sample sizes for these cohorts, differences in allele frequencies, and differences in epidemiological and/or environmental influences between the ancestry groups.
For example, the disease signatures identified in the Sano GOLD cohort may have reduced effect sizes in non-white European cohorts due to an increased frequency of ‘actively protective’ signatures in those populations, i.e., one or more SNP genotypes that wholly or partially mitigate the disease associations of a set of ‘causative’ disease signatures [50]. The combinatorial analysis in this study focused only on causative disease signatures and did not include any analysis of protective signatures30. Incorporating actively protective features into the set of disease signatures should increase their predictivity for identifying ‘high-risk’ patients and improve reproducibility statistics.
These results highlight a pressing need to identify large, diverse, well-phenotyped cohorts of long COVID patients. Many long COVID specific datasets such as Sano GOLD are comprised predominantly of patients with white European ancestry. In contrast, All of Us includes a highly diverse patient cohort, but lack of reliable data identifying which participants have a history of long COVID prevents us from reliably obtaining sufficient sample sizes to conduct a combinatorial analysis aimed at identifying novel disease signatures.
Having access to larger and more diverse populations with a confirmed diagnosis is essential to enabling primary analysis of disease risk and protective factors within these ancestry cohorts and adding to our understanding of the factors underpinning disease in those populations. In turn this would also help us build more inclusive and transferrable disease risk models. Notably, combinatorial analysis of diverse long COVID patient cohorts could potentially identify disease signatures that were not detected in predominantly white European cohorts due to low relative case frequencies, but which have greater frequency and importance for disease biology in other patient cohorts. Such signatures could be used to better estimate patients’ relative susceptibility to developing long COVID.
Evaluating the output of the PrecisionLife combinatorial analysis platform
We observed high rates of reproducibility among disease signatures derived from all the Sano GOLD cohorts and showed that these rates of disease signature reproducibility were strongly correlated with the frequency of signatures in the original study cohort. We observed slightly higher overall rates of reproducibility in the Severe and Fatigue Dominant cohorts which have fewer high case frequency disease signatures relative to the broader ‘General’ long COVID cohort.
Rates of reproducibility were highest for disease signatures comprised of four or five SNP genotypes, suggesting that combinatorial genetic interactions play an important role in the biology of long COVID. This also provides supporting evidence for the combinatorial analytic approach’s ability to detect a broad and clinically informative set of genetic disease associations in otherwise intractable complex diseases.
The predictive value of common vs rare signatures
In contrast to these mid/high case frequency signatures, when analyzing the entire set of disease signatures from the original analyses including low frequency signatures, only 34–44% were consistently correlated with long COVID prevalence. This implies that rarer signatures may replicate between populations more poorly, an observation that is consistent with similar findings in GWAS and polygenic risk score studies [51,52,53,54,55]. There are several explanations for this observed correlation between signature frequency and reproducibility rates.
First, statistical power is proportional to sample size, which is already limited in the reproducibility analysis due to the very small number of confirmed long COVID patients in AoU. Signatures with frequencies below 5% are expected to occur in 21 or fewer AoU cases. This small sample size results in large variance in expected rates of reproducibility under the null model and a high probability of observing odds ratios less than one due to random sampling even when signatures are biologically relevant to disease.
Second, we demonstrated that reproduction rates for low frequency signatures with moderate-to-high true odds ratios are most strongly negatively affected by the likely inclusion of patients with long COVID in the control cohort.
Third, due to the high case:control skew (1:10) in our dataset, rare signatures were often more likely to be negatively correlated with disease under the null model. In the most extreme scenario, a signature that occurs in one person in the dataset is 10 times more likely to randomly occur in a control (resulting in a negative odds ratio) than a case (resulting a positive odds ratio). This bias caused the mean numbers of signatures that randomly exhibit odds ratios above 1 in the null model permutations to range between 41 and 46% – below the 50% expectation for a balanced dataset.
Fourth, rare signatures appeared to be more reflective of subpopulation structure in the original Sano GOLD dataset. Including genetic principal components as covariates resulted in 4% fewer high-frequency signatures (i.e., those that occur in > 5% of total cases) that are positively correlated with long COVID, relative to a logistic regression that did not include covariates for population substructure. In contrast, including genetic principal components in the analysis resulted in 52% fewer replicating low-frequency signature (i.e., those that occur in
Finally, more complex disease signatures (i.e., those comprised of 4 or 5 SNP genotypes) generally occur at lower frequencies in the population simply because there are more possible genotype combinations for a larger set of SNPs. The risk of overfitting to a dataset is known to increase with tree depth when applying tree-based machine learning algorithms [56] and the same potentially holds true for higher layer disease signatures derived from a layer-based mining approach. Applying a frequency filter therefore potentially mitigates the impacts of false positive SNPs by removing higher-order signatures.
We found no evidence, however, that increased signature complexity was associated with reduced reproducibility among high-frequency signatures. Rather, overall reproducibility rates were highest for 4-SNP and 5-SNP signatures relative to the small number of 2-SNP signatures. We also did not observe a correlation between signature complexity and reproducibility rate among low-frequency signatures. These results suggest that outputs of the combinatorial analyses of the Sano GOLD cohorts were not excessively overfitted to the original datasets and that presence of any false positive component SNP genotypes does not significantly affect the overall association with disease.
Although the results of this analysis suggest that false positive component SNP genotypes do not have a major effect on signature reproducibility, we could potentially improve the effect sizes and predictivity of these signatures by using AoU to further refine the set of signatures. This step entails testing each signature individually and removing any component SNP genotype that does not enhance the signature’s association with disease in AoU. We have not included any refinement analysis in this study because it can potentially overfit the new set of signatures to the training dataset (AoU). A third cohort of long COVID patients would be required to properly evaluate the improvement in disease signature reliability that results from this refinement process.
Limitations of analysis
Reliably identifying which patients in AoU have a history of long COVID is currently challenging. We relied on ICD-10 coding, which is known to be inconsistently and inaccurately applied, to identify known cases. As noted above, published estimates of long COVID prevalence in the United States range between 6.9% to 14%, yet fewer than 0.2% of individuals in AoU have ICD-10 codes associated with long COVID.
This suggests that many long COVID patients have not been assigned the appropriate ICD-10 code. As a result, more than 10% of the controls in our AoU study cohort potentially could represent misclassified cases with unreported long COVID. We have, however, noted in similar studies of highly heterogenous diseases that constraining diagnostic criteria in this manner (at the cost of potentially missing true cases) is more effective than including a larger number of poorly diagnosed (potentially true negative) patients into the case group.
This type of phenotypic misclassification in datasets will generally weaken the observed effect sizes by artificially inflating the similarity between cases and controls [57]. This behavior is potentially problematic for reproducibility analyses, as the dilution of signal decreases the statistical power of the analysis [58]. For example, phenotypic misclassification increases the probability that a signature that is biologically correlated with increased disease risk will nonetheless exhibit an odds ratio less than 1 due to random sampling effects.
We therefore expect that the high degree of phenotypic misclassification in our dataset will have worked to reduce the overall rates of observed signature reproducibility. As such, the reproducibility statistics presented in this paper probably represent a low-end estimate of the true reproducibility rate. Better diagnostic criteria and the use of harmonized surveys of patients’ self-reported symptoms would help pool patient datasets and compare results across cohorts.
We noted that 41% of signatures (3725/9068) could not be tested due to missing SNPs in AoU. These were predominantly SNPs that were removed from the AoU dataset before its release, presumably due to data quality issues [59]. The removal of these SNPs skew the reproducibility analysis toward slightly more common signatures, as signatures comprised of many SNPs tend to have lower frequencies in the population and are also more likely to include a missing SNP. Because they are enriched in rarer signatures, inclusion of these missing SNPs may have resulted in slightly weaker overall reproduction rates. They would not be expected to have a significant effect on the reproduction statistics for more common variants, however, and may have resulted in stronger p-values due to larger sample sizes of signatures tested.
There was much more heterogeneity and diagnostic uncertainty about the General Cohort in the original Sano GOLD analysis relative to the more well-defined Severe and Fatigue Dominant cohorts. This included small sets of long COVID ‘patients’ self-reporting no prior COVID-19 infection, and others reporting improvements in their health post COVID-19 infection.
It was therefore unsurprising that this more heterogeneous cohort was associated with more signatures in the Sano GOLD study, consistent with the assumption that a wider variety of phenotypes will be linked to more genes. This large number of signatures is problematic when attempting to statistically validate the replication of individual disease associations from the General cohort due to the need for more stringent FDR correction. As a result, none of the individual General cohort signatures statistically replicated, even though these include signatures that have stronger p-values than the signatures in the Fatigue Dominant and Severe cohorts, which did statistically replicate. A much larger cohort is required to overcome this multiple testing burden and allow for statistical validation of individual signatures from the General cohort.
Applications for healthcare
The identification of a set of genetic signatures that are consistently associated with increased prevalence of long COVID offers many opportunities for improving treatment of this poorly understood but highly prevalent and debilitating disease.
Firstly, although we have insufficient power to validate the full set of individual signatures in AoU, demonstrating that reproducing signatures are significantly enriched in a second dataset provides important confirmatory evidence of the original findings of the combinatorial analytics approach. To provide insights into potential drug therapies for long COVID, we further tested whether the signatures associated with novel drug targets and their related drug repurposing candidates are significantly correlated with increased long COVID prevalence in AoU. 27/30 (90%) of the genes represented in the > 5% disease signatures and 11 out of the 13 drug repurposing candidates identified in the original study were reproduced in this study. This lends weight to their prioritization in clinical efficacy trials especially for those with generic drugs.
Controlled open-label studies of similar design to the RECOVERY trial in Covid-19, which rapidly identified dexamethasone as an effective frontline therapy [60], can be undertaken on this set of generic drugs, benefiting from the additional evidence that one or more selected therapies is more likely to help the subset of patients who have those mechanisms’ disease signatures in their genetic makeup.
Secondly, we can use the insights into disease biology that are reflected by the reproducing disease signatures to construct a combinatorial risk score that evaluates an individual patient’s relative genetic susceptibility towards developing long COVID. Although genetic risk scores are not strictly diagnostic, especially in a pathogen triggered disease, they have substantial potential to be used by physicians for differential triage, i.e., to rapidly gauge the relative likelihood of different diagnoses when presented with ambiguous or indistinct symptoms and refer patients and/or select treatment options accordingly. As the utilization of large-scale COVID-19 testing fades, alternative tests that can help differentiate patients with long COVID from patients with other illnesses with similar symptoms will become increasingly useful in healthcare settings.
Constructing a combinatorial risk score from disease signatures is a more complex challenge than a conventional polygenic risk model—the latter assumes that all features (SNPs) act independently, whereas combinatorial disease signatures are often inherently correlated due to the sharing of SNP genotypes. Machine learning approaches can disentangle this complexity and non-independence and combine features such as disease signatures and their component SNP genotypes into a single predictive model. Although the small sample size of the AoU dataset is sufficient to train a combinatorial risk score using machine learning, a third (currently unavailable) independent dataset would be required to properly evaluate the relative increase in long COVID prevalence between subsets of patients flagged as having high and low genetic susceptibility.
Finally, the set of replicating disease signatures can be used to mechanistically stratify patients based on the causative etiologies most likely associated with their form of long COVID. This first entails assigning disease signatures to one or more mechanism-of-action (MoA) cluster(s) based on the gene(s) associated with the component SNP genotypes. We can then assess whether a patient has a significant excess or lack of disease signatures associated with a given MoA relative to the distribution of signature counts in the larger long COVID community. In essence, this mechanistic stratification tool is comprised of multiple combinatorial risk scores, each for a different set of mechanism related disease signatures. This can provide insight in the clinic into the selection of therapies that are matched to a patient’s personal genetic makeup.
Unlike standard risk scores, which can be used to inform public health applications but provides more limited utility for personalized precision medicine [61], a mechanistic stratification tool would potentially ultimately enable healthcare practitioners to identify individualized treatment regimens including single- or multi-drug therapies that are most likely to generate a positive outcome for a given patient. In the case of long COVID these mechanistic insights also have other potential applications, as the Taylor et al. (2023) combinatorial analysis also found evidence for substantial overlap in disease biology between long COVID and myalgic encephalitis/chronic fatigue syndrome (ME/CFS)30.