The A20 acts as a critical negative regulator of the NF-κB signaling pathway by suppressing inflammatory cascades through its dual enzymatic domains(OTU and ZnFs) [7, 10, 31]. Beyond NF-κB, emerging evidence highlights its regulatory roles in the JNK and JAK-STAT pathways, underscoring its broad influence on immune homeostasis [8]. Expressed ubiquitously in immune and epithelial cells, A20 modulates diverse processes, including apoptosis, inflammation, and oncogenesis [9]. Structural analysis reveals that the OTU domain deubiquitinates key mediators of the IKK complex, dampening NF-κB activation, while the ZnF domain facilitates RIPK1 ubiquitination and degradation, further curbing inflammatory signaling [8, 11]. Loss of A20 function disrupts this regulatory balance, leading to unchecked NF-κB activation and predisposing individuals to autoimmune and autoinflammatory disorders [12, 13].
Patients with HA20 demonstrate a broad clinical spectrum characterized by multisystem involvement [20]. The predominant manifestations include recurrent oral aphthosis (present in 68% of cases), genital aphthosis (37%), periodic fever patterns (48%), cutaneous eruptions (41%), and gastrointestinal pathology manifesting as abdominal pain, diarrhea, or hematochezia (39%) [14, 15, 20]. Lymphadenopathy and recurrent respiratory infections are frequently observed comorbidities. Notably, approximately 30% of patients develop autoimmune sequelae ranging from Hashimoto’s thyroiditis to systemic lupus erythematosus [16], and rheumatological complications such as arthritis [17]. Less common but clinically significant associations include interstitial pneumonia, neuroinflammatory manifestations, and hematological malignancies like Hodgkin’s lymphoma [14, 15, 17].
Geographical variation in phenotypic expression has been well-documented. East Asian cohorts predominantly exhibit periodic fever syndromes of unknown etiology (70.4% vs. 37.3% in non-Asian populations, P 14]. This regional disparity extends to autoimmune manifestations, with East Asian patients demonstrating lower incidence rates of autoimmune diseases (29.2% vs. 58.8%, P
The molecular pathogenesis of HA20 centers on TNFAIP3 mutations disrupting A20’s dual regulatory domains [18]. Since the initial 2016 characterization, 75 genetic variations at the TNFAIP3 locus have been cataloged [19,20,21,22], and 12 cases of chromosome 6q23 segmental deletions causing complete A20 deficiency [23, 24]. These large deletions (median size 3.35 Mb) typically abrogate both OTU and ZnF domains, resulting in constitutive NF-κB activation through loss of deubiquitinase and ubiquitin-binding functions. Clinically, complete A20 deficiency correlates with early disease onset (median age 4 months), severe multiorgan involvement, and neurodevelopmental abnormalities (4.2% incidence) [24,25,26]. Our Patient 2 exemplifies this severe phenotype, presenting at 2 months with pan-gastrointestinal ulceration and progressing to joint destruction, highlighting the prognostic challenges in this subgroup requiring vigilant neurological monitoring. Patient 2 harbors a 2.18 Mb heterozygous deletion on chromosome 6 (6q23.3), resulting in the loss of 13 protein-coding genes, notably TNFAIP3, PEX7, IL20RA, and IL22RA2. The TNFAIP3 deletion, causative of HA20, underlies the core Behçet’s-like phenotype characterized by fever, abdominal pain, diarrhea, hematochezia, skin eruption, Oral aphthosis and genital aphthosis, intestinal ulcers, and arthritis. The observed arthritis may potentially relate to PEX7 deletion [27], while deletions in IL20RA and IL22RA2 might exacerbate inflammatory dysregulation or skin eruption [28].
Mutation-specific genotype–phenotype correlations continue to evolve. Frameshift/nonsense mutations predispose to immune dysregulation with recurrent infections and cytopenias, while missense variants associate with cutaneous vasculitis [18, 24]. Domain-specific analyses reveal OTU domain mutations preferentially link to Behçet’s-like phenotypes, whereas combined OTU + ZnF disruptions correlate with musculoskeletal complications, suggesting that the ZnF domain may play an important role in the pathogenesis of musculoskeletal diseases in HA20 patients [29]. ZnF7 domains deletion have been reported in mouse to result in a spontaneous inflammatory disease, but not ZnF4 deletion [30]. However, recent meta-analyses challenge these associations, emphasizing the need for larger cohort studies to resolve current controversies in phenotypic stratification [12, 24].
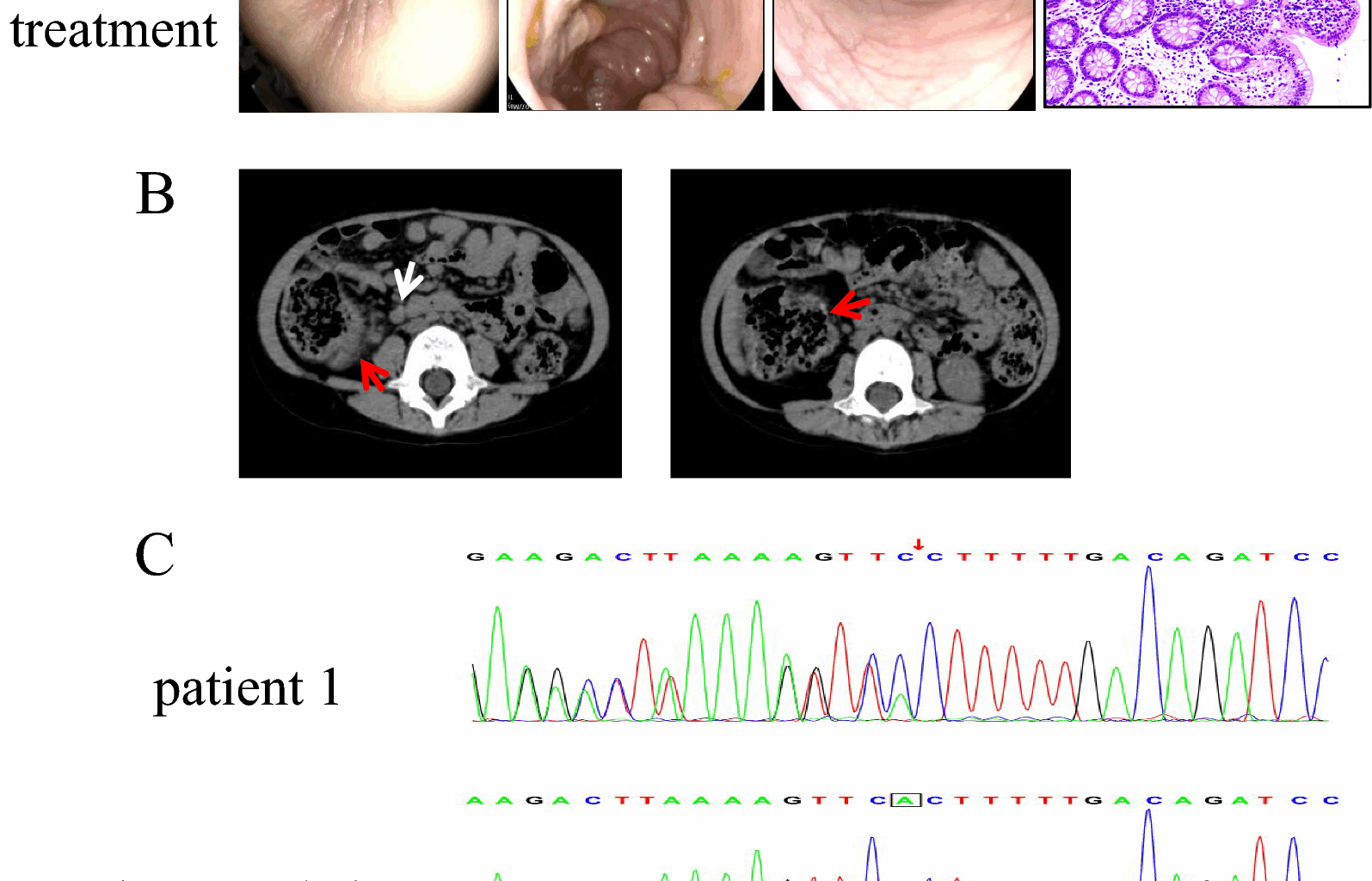
In this study, Patients 1 and 4 both exhibited frameshift mutations at previously unreported sites. These distinct mutations correlated with differing clinical presentations. Patient 1 carried a TNFAIP3 mutation (c.866delA), leading to p.His289Profs*3. This highly pathogenic frameshift mutation disrupts part of the OTU domain and causes complete loss of all zinc finger domains (ZnFs). The resulting severe impairment of A20 function manifested as significant gastrointestinal symptoms and recurrent infections, with suboptimal therapeutic response. Patient 4, however, harbored a TNFAIP3 variant (c.1243_1247del; p.Asn416Thrfs*11). This mutation preserved the OTU and ZnF1, resulting only in the loss of ZnF2-ZnF7. The clinical presentation featured predominant recurrent infections and milder gastrointestinal symptoms. The observed liver injury and rash support the proposed role of ZnF domains in regulating organ-specific inflammation [24]. These may also suggest an association between OTU domain alterations and Behçet’s-like phenotypes [29]. Patient 3 presented a nonsense mutation (c.133C > T; p.Arg45Ter), causing A20 truncation. This abolished both the OTU domain’s deubiquitinase activity and the ubiquitin ligase activity of all zinc fingers. Remarkably, after nine years of follow-up, this patient has achieved complete clinical and endoscopic remission. These findings show both consistency and contradictions with Chen et al.’s research [29], underscoring the disease’s complexity and the critical need to expand the catalog of documented TNFAIP3 variants. This study describes four TNFAIP3 variants, three of which are novel (not previously reported). Our primary contribution lies in enriching the existing repository of TNFAIP3 variants. At present, definitive genotype–phenotype correlations cannot be established based on this cohort.
The establishment of definitive diagnostic criteria for HA20 remains an unmet clinical need, complicated by its phenotypic heterogeneity and lack of pathognomonic laboratory findings. During acute flares, elevations in ESR, CRP, proinflammatory cytokines (e.g., IL-6), and autoantibodies may occur, though these markers merely reflect systemic inflammation rather than providing diagnostic specificity. In our cohort, all four patients demonstrated anemia (likely secondary to chronic gastrointestinal bleeding) with Patient 2 exhibiting severe anemia requiring hematological intervention. Joint MRI in this patient revealed compensatory erythroid hyperplasia. While immune dysregulation is central to HA20 pathogenesis [31]. Evidenced by T/B lymphopenia in Patients 1 and 2 potentially reflecting chronic inflammation-induced cytotoxicity, these cellular alterations lack diagnostic specificity. Experimental evidence implicates TNFAIP3 deficiency in immune tolerance breakdown, with dendritic cell-specific knockouts developing SLE-like phenotypes [32] and B cell-specific deletions causing spontaneous autoantibody production [33]. Such mechanisms may explain the observed complement activation (elevated C3/C4) and cytokine-driven T/B cell dysfunction in HA20 patients [31, 34]. Genetic confirmation through whole-exome or genome sequencing has become the diagnostic gold standard. We recommend prompt genetic evaluation for patients presenting with: (1) Early-onset disease (
Therapeutic strategies remain empirical, reflecting the disease’s variable clinical trajectories. First-line corticosteroids, though effective for acute control, face adherence challenges due to growth-related complications in pediatric populations. Conventional immunosuppressants (methotrexate, azathioprine) typically require glucocorticoid co-administration for sustained remission [8]. Emerging biologic therapies-including TNF-α inhibitors (infliximab), IL-1/IL-6 pathway antagonists (anakinra, tocilizumab), and JAK inhibitors (tofacitinib)—offer targeted modulation of hyperinflammatory responses, particularly in refractory cases [35]. Critical considerations for treatment personalization include: Developmental stage-adjusted dosing regimens; Predominant pathophysiological drivers (autoinflammatory vs autoimmune dominance); Mutation-specific functional impacts (OTU vs ZnF domain perturbations). In this report, All patients received EEN and thalidomide, a regimen effective in mild-moderate cases. Patient 1’s switch to infliximab after thalidomide intolerance highlights TNF-α’s central role in HA20 pathogenesis, particularly in OTU domain defects. Conversely, Patient 2’s refractory disease may reflect irreversible NF-κB hyperactivation due to complete A20 loss, necessitating combination biologics (e.g., IL-1/IL-6 inhibitors) or JAK inhibitors. The variability in mucosal healing complete in Patient 3 versus persistent aphthosis in Patient 2 suggests that residual A20 function, dictated by mutation type, influences tissue repair capacity.
This study’s small sample size and retrospective design limit generalizability. Longitudinal follow-up (4–10 years) revealed evolving phenotypes, yet longer observation is needed to assess late complications (e.g., malignancy). Furthermore, functional studies are imperative to clarify how specific mutations dysregulate A20’s interactions with RIPK1 or TRAF6. Multicenter cohorts and standardized treatment protocols are essential to validate genotype-driven therapies and establish prognostic biomarkers.