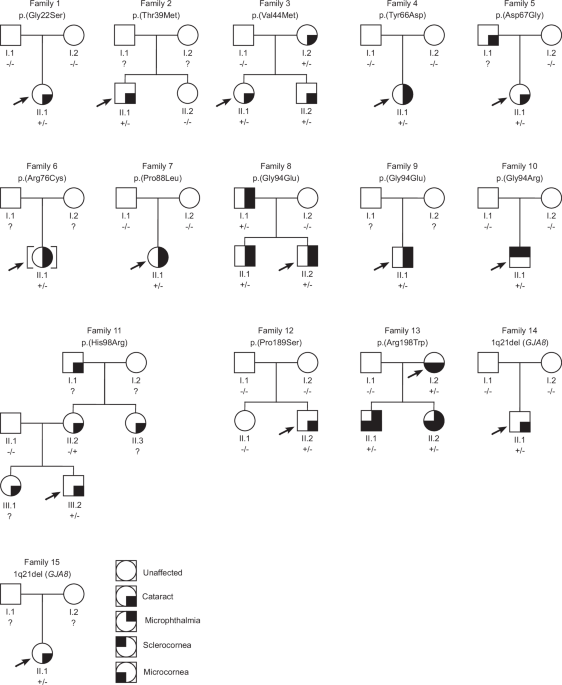
Here, we report variants affecting GJA8 in 22 individuals from 15 new families, including 11 missense variants predicted pathogenic/likely pathogenic, one missense variant of uncertain significance, and two heterozygous 1q21 microdeletions of uncertain significance. Segregation analyses, available in ten families, demonstrated that six variants were de novo and four variants followed an autosomal dominant inheritance pattern. Of the 12 missense variants identified, one is novel [p.(Tyr66Asp)], another [p.(Thr39Met)] is very rare in public genomic databases and is not linked with any condition, while the remaining 10 have been previously reported in cases with eye anomalies. Together, our data expand the spectrum of GJA8 variants, and help to delineate genotype-phenotype relationships.
Pathogenic variants in GJA8 were initially described in individuals with isolated childhood cataract [10]. As a result, studies investigating this gene have mainly focused on single families or cohorts with congenital cataracts [15]. More recently, it emerged that individuals with GJA8 variants can also display more severe lens anomalies (aphakia) or broader ocular phenotypes, including microphthalmia and anterior segment anomalies, typically in combination with cataract, and occasionally with extraocular features [11, 12, 16]. Our international study broadened the analysis to include subjects with a range of developmental eye anomalies. Six of the 13 families with missense GJA8 variants identified herein presented with isolated cataract, without microphthalmia (families 1, 2, 3, 5, 11 and 12). Six families exhibited both cataract and microphthalmia (families 4, 6, 7, 8, 9 and 13), and one presented with microphthalmia and sclerocornea (family 10). Although it is not possible to draw conclusions about the relative frequency of isolated cataract versus cataract with microphthalmia in individuals with GJA8 variants in this study, due to recruitment biases in the four cohorts, our screening confirms that alterations in this gene represent an important cause of microphthalmia. Interestingly, the proband in family 10 was diagnosed with microphthalmia and sclerocornea, but not cataract. While lens abnormalities can be ascertained using ultrasound in the presence of sclerocornea, this was not performed during the clinical assessment of family 10 and the proband was unavailable for further examination to determine the presence and/or characteristics of the lens. This illustrates how detailed phenotyping, including ultrasound, can potentially contribute to a better understanding of the range of features associated with GJA8 variants and direct genetic testing. In addition to cataract, microphthalmia and sclerocornea, all affected individuals in families 3, 4, 5, 6, 8, 11 and 13 displayed other associated ocular features, including glaucoma, nystagmus, esotropia and amblyopia. However, many of these were secondary to the early onset cataract, structural eye anomalies and/or surgical interventions.
Of the 12 missense variants identified in this study, 10 have been previously reported in individuals with eye anomalies, allowing some genotype-phenotype correlations. The variants p.(Gly22Ser), p.(Val44Met), p.(Asp67Gly) and p.(Pro189Ser) were reported in individuals with cataract, but no structural eye anomalies, both in our cohort and previous studies [17, 27,28,29,30,31,32]. The variant p.(Gly94Arg), has been previously reported in four independent cases; the first presented with bilateral sclerocornea, microcornea and rudimentary lenses [12], the second with bilateral sclerocornea, microphthalmia, colobomas and congenital aphakia [11], the third with bilateral sclerocornea and congenital aphakia [13], and the fourth with microphthalmia and sclerocornea [33]. Here, we identified the same variant in a fifth case (family 10), also presenting with bilateral microphthalmia and sclerocornea. While the lens phenotype of the proband in family 10 was not assessed, this additional case strengthens the link between p.(Gly94Arg) and sclerocornea. Interestingly, sclerocornea occurring with aphakia/abnormal lens phenotype is also associated with biallelic variants in FOXE3 [34]. Therefore, our findings indicate that genetic screening of individuals with sclerocornea and lens abnormalities, including aphakia, should involve GJA8 in addition to FOXE3. Families 8 and 9 carry an alternative substitution affecting residue 94, p.(Gly94Glu). Interestingly, this change has been reported in a proband and mother with microphthalmia [14] and an individual with sclerocornea and microcornea [12], whereas families 8 and 9 in this study exhibited cataract and microphthalmia without sclerocornea. This demonstrates inter-familial phenotypic variability associated with this GJA8 variant, with significant implications for clinical assessment and genetic counselling.
Such inter-familial phenotypic variability has been previously reported for other GJA8 variants [11], and is observed for additional alterations in this study. For example, the probands carrying the variants p.(Arg76Cys) and p.(Pro88Leu) (families 6 and 7, respectively) exhibited congenital cataract and microphthalmia. However, previously reported individuals who carried these variants displayed isolated congenital cataracts [17, 35]. Furthermore, intra-familial phenotypic variation was also evident. In family 13, the affected mother and her two children carried the p.(Arg198Trp) variant and displayed cataract and microcornea features. However, her two children additionally manifested bilateral microphthalmia, microcephaly and developmental delay. The chromosome 2q14.2 duplication, previously reported in this family [26], is unlikely to explain the differences in phenotypes since all the affected individuals share this variant; no other variant was identified to explain the microcephaly. Interestingly, the same missense variant p.(Arg198Trp) was identified in a family with congenital cataract and microcornea, without any additional ocular or extraocular features [36], further highlighting inter-familial variability. Variation in phenotypic features is often attributed to environmental, genetic and local stochastic factors [37]. Six families with missense GJA8 variants in the present study (families 6, 7, 9, 10, 11 and 13) displayed additional systemic anomalies, some explained by additional genetic variants. Of these, family 6 with tooth agenesis and family 11 with high myopia and congenital deafness, had additional variants in WNT10A and COL11A1, respectively, accounting for their extraocular features. Similarly, in the study by Ceroni et al. (2019), the systemic anomalies in some individuals with pathogenic GJA8 variants were explicable by additional genetic variants [11]. Therefore, while no additional pathogenic variants were reported in the remaining individuals with systemic anomalies in the present study, further genetic analyses, especially of non-coding genomic regions, may be vital to unravel these additional features.
In addition to glycine 94, our study highlights two more mutational hotspots, threonine 39 and tyrosine 66. Two different alterations affecting threonine 39 have been reported, p.(Thr39Arg) and p.(Thr39Lys). The variant p.(Thr39Arg) has been described in two unrelated individuals, one with congenital cataract, microcornea and iris hypoplasia [38] and the other with congenital cataract, microphthalmia and corneal opacification [11]. The individual with p.(Thr39Lys) displayed microphthalmia, microcornea, cataract and anterior chamber anomalies with some systemic manifestations including neurodevelopmental delay, decreased body weight, and short stature (ClinVar: SCV004183581). We report a new third substitution of this residue, p.(Thr39Met), in an individual with isolated bilateral congenital cataract (family 2). This same variant is reported in one individual in the most recent release of gnomAD database (v4.1.0), but their ocular phenotype is not specified. Since this individual is from the UK Biobank, a population-based dataset that includes individuals who might have health conditions [39], it is possible that they have eye anomalies. Moreover, a recent study identified the same p.(Thr39Met) variant naturally occurring in cavefish with eye anomalies, including microphthalmia [40], providing further evidence that the variant may be causal. Similarly, we report a novel change of tyrosine 66, p.(Tyr66Asp), in an individual with bilateral cataract and microphthalmia (family 4). Three different pathogenic changes affecting this residue are reported in ClinVar [p.(Tyr66Cys) (SCV001379194), p.(Tyr66His) (SCV000952358) and p.(Tyr66Ser) (SCV001219479)], all in individuals with developmental cataract, without reported microphthalmia. Together, these data support the hypothesis that different substitutions at threonine 39 and tyrosine 66 can lead to developmental eye anomalies, predominantly cataract, with additional features perhaps influenced by the nature of the variant, but also other genetic and/or environmental factors.
Our study also identified two families with 1q21 microdeletions affecting GJA8. The impact of microdeletions affecting GJA8 remains uncertain, due to several reported carriers having no apparent disease phenotype [11, 41, 42]. Interestingly, in one study, re-examination of a reportedly unaffected carrier of a 1q21 microdeletion revealed subtle lens opacities and a patent ductus arteriosus, which may be unrelated [43]. In contrast, in a family described by Ceroni et al. (2019) (family 15 [11]), in which the proband carried a heterozygous de novo 1q21 microdeletion involving GJA8, a pathogenic FZD5 variant was subsequently identified [Individual 7 [42]], explaining their ocular phenotype. However, the proband who carried both the 1q21 microdeletion and FZD5 variant displayed a more severe disease phenotype than her father and paternal cousin who only carried the FZD5 variant. This suggests that the 1q21 microdeletions may either confer susceptibility to disease or contribute to its severity. Therefore, future studies of gene-gene interactions relevant to 1q21 microdeletions may unravel pathogenic mechanisms.
The expanding number of GJA8 missense variants associated with severe ocular features contributes to our knowledge of the residues crucial for GJA8 activity. It was previously noted that cataract-associated GJA8 variants clustered in the transmembrane domains, specifically between TM1 and TM2, while severe phenotypes, including microphthalmia, were more likely to be associated with alterations occurring in the extracellular and cytoplasmic loops [11, 44]. Our findings of cataract associated with the variants located in TM1 [p.(Gly22Ser) and p.(Thr39Met)], and microphthalmia and sclerocornea associated with variants in ICL [p.(Gly94Glu) and p.(Gly94Arg)] support this view. However, in our cohorts, p.(Arg76Cys) and p.(Pro88Leu), both located in TM2, were also found to be associated with severe ocular phenotypes including microphthalmia (families 6 and 7). This suggests that the phenotype associated with GJA8 variants might not be domain-specific, but rather dependent on the particular residue and/or substitution.
The complex ocular phenotypes displayed by individuals with GJA8 variants could also partly be explained by the expression pattern of GJA8 within the eye. GJA8 is expressed widely throughout the lens [8], and also in the cornea [45, 46], indicating an additional role in corneal gap junctions. A related connexin, GJA3 (Cx46), is predominantly expressed in lens fibres, and individuals with GJA3 variants display cataract-only phenotypes. These differences are recapitulated by Gja3 and Gja8 mouse models where Gja3-null mice had cataract but normal eye size [47], while Gja8-null mice developed cataracts earlier and exhibited smaller lenses and microphthalmia [5, 6]. Mice/humans with variants in other cataract-associated genes, such as crystallins can also manifest with complex features including corneal anomalies, iris hypoplasia and microphthalmia [(CRYAA [48]; CRYBB1 [49]; CRYBA4 [50]]. This further indicates that several cataract-associated genes, including GJA8, may have broader roles in ocular growth and development.
In conclusion, we report 22 individuals from 15 new families with GJA8 variants identified from a large international multicentre cohort of individuals with developmental eye anomalies. We highlight that GJA8 variants represent an important source of genetic diagnoses not only for individuals with early onset cataract, but also for individuals with developmental eye anomalies, including microphthalmia and sclerocornea. Our data illustrates GJA8 mutational hotspots, and significant inter- and intra-familial variation associated with GJA8 variants. Further genotype-phenotype studies of individuals with rare developmental eye anomalies will be vital in defining the role of GJA8 and other genes in their pathogenesis. This, in turn, will improve diagnosis, counselling of families, and provide insight into future therapies.