Structural and photo-physical characterizations
The crystal structure of triclinic CuSeO3 with space group P1 is illustrated in Fig. 1a. Two inequivalent Cu sites are involved in one individual CuSeO3 unit cell. In the first site, the Cu atom is bonded to five O atoms forming corner sharing CuO5 trigonal bipyramids. In the second site, the Cu atom is bonded to four O atoms in a square co-planar geometry. There is no direct bond between Cu and Se atoms. Their linkage is mediated via O, where each Se atom binds with three O atoms to form a trigonal non-coplanar geometry (SeO32-). It should be mentioned that the successful synthesis of triclinic CuSeO3 had only been reported until 202053, by Bera and Chakraverty in their research to develop helimagnetic spin structures toward spintronic applications. However, in their work, the obtained CuSeO3, although light green-colored, was recognized as a Mott insulator with the band gap evaluated at 3.9 eV.
Fig. 1: Characterizations of CuSeO3-x.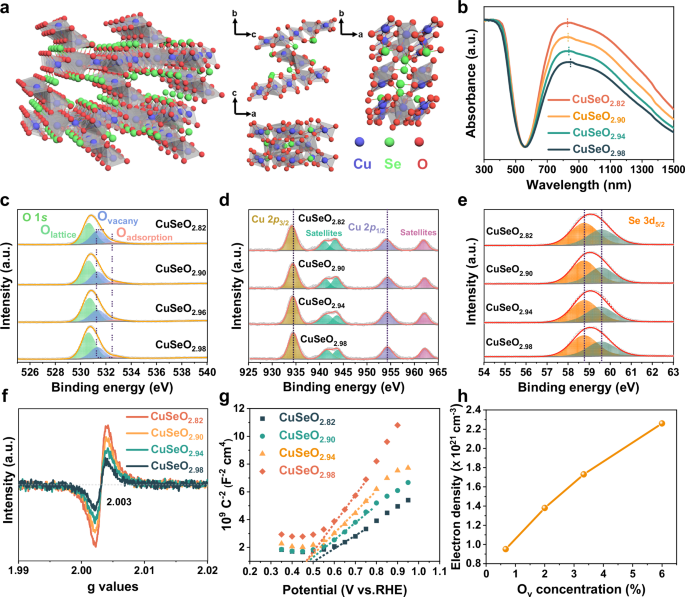
a Illustration of the crystal structure. b UV-Vis-NIR DRS spectra. c O 1 s, d Cu 2p, and e Se 3 d XPS spectra. f EPR spectra. g Mott-Shottky plots. h Correlation between Ov concentration and free electron density in CuSeO3-x. Source data for b–h are provided as a Source Data file.
In our study, the triclinic CuSeO3-x crystals featuring intensive LSPRs absorption in NIR region were accessed using a similar method via a hydrothermal process with modifications (for example, sharply reducing the reaction time from 72 h to 6 h)53. CuCl2•2H2O and Na2SeO3 were adopted as the precursors and their molar ratio was finely tailored from 1:1 to 1:0.97, 1:0.94 and 1:0.91. According to the results of inductively coupled plasma mass spectrometry (ICP-MS), the chemical compositions of thus prepared samples are CuSeO2.98, CuSeO2.94, CuSeO2.90 and CuSeO2.82, correspondingly (Table S1). As revealed in Fig. S1, XRD patterns of these CuSeO3-x are well matched with the structural phase of triclinic CuSeO3 (JCPDS 48-0375) without any impurities. However, continuing varying the molar ratio between Cu and Se precursors would lead to the generation of a second phase. Transmission electron microscopy (TEM) image in Fig. S2 indicates that the formed triclinic CuSeO3-x is composed of irregular bulk crystals. Very interestingly, it was found that these series samples manifest a bright green color (Fig. S3). Such observation is in good agreement with the UV-visible-NIR diffuse reflectance spectra (DRS) as shown in Fig. 1b. One can see that in addition to the interband transition absorption below ~600 nm, there is a strong and broad band across the long-wavelength visible and NIR range (~700–1500 nm) centered around 830 nm.
It is well acknowledged that copper chalcogenide nanocrystals, such as Cu2-xSe, exhibit notable NIR absorption arising from LSPRs due to a high density of Cu vacancies40,46,47,48. In our case, the stoichiometric ratio between Cu and Se determined by ICP-MS demonstrates that the remarkable absorption beyond band edge observed for CuSeO3-x should not follow the same mechanism as Cu2-xSe. Alternatively, the obvious deficiency of oxygen atoms in these samples suggests that the existence of anionic oxygen vacancies is probably an important factor. To validate this assumption, X-ray photoelectron spectroscopy (XPS) measurements were carried out over the prepared CuSeO3-x. From Fig. 1c, the O 1 s spectra can be deconvoluted into three components: the peak at 531.6 eV attributed to oxygen vacancy, and other two peaks at 530.5 and 532.3 eV associated with lattice oxygen and surface adsorbed species (e.g., OH), respectively49,54,55,56. As denoted by the alteration in peak areas, decreasing the addition amount of Na2SeO3 during synthesis resulted in a gradual increase in the concentration of oxygen vacancies, consistent with the conclusion derived from ICP-MS. For Cu 2p spectra (Fig. 1d), the two peaks at 934.3 and 954.2 eV accompanied by evident satellites are characteristic of Cu2+. It can be noticed that along with the increment in Ov concentration, the Cu 2p peaks showed a slight shift to lower binding energies indicative of the enhanced electron density around Cu. Furthermore, the Se 3 d bands at 58.8 and 59.6 eV uncovered that in CuSeO3-x the valence state of Se is +4 (Fig. 1e)57,58. The presence of oxygen vacancies was further verified by electron paramagnetic resonance (EPR) characterizations. In Fig. 1f, the same variation trend as XPS spectra was observed, supporting that the concentration of oxygen vacancies in CuSeO3-x can be readily modulated by adjusting the molar ratio between Cu and Se precursors.
Previous investigations have established that a high charge carrier density on the order of 1021 cm−3 is essential to give LSPRs response in the NIR spectrum46,59. To determine the carrier density in CuSeO3-x, Mott-Schottky measurements were conducted on the basis of Eq. (2):
$$\frac{1}{{C}^{2}}=\frac{2}{e{A}^{2}{\varepsilon }_{0}{\varepsilon }_{A}N}\left(V-{V}_{{fb}}\right)$$
(2)
where C is the charge layer capacitance, e (1.602 × 10−19 C) is the elementary charge, A (1 cm2 in our case) is the surface area, ε0 (8.854 × 10−14 F cm−1) is the electrical permittivity in vacuum, εr is the relative dielectric constant for CuSeO3-x (measured to be 6.05 through four probe method, Table S2), N is the charge carrier density, V is the applied potential and Vfb is the flat-band potential60,61,62. As displayed in Fig. 1h, the positive slopes of Mott-Schottky plots indicate the n-type conductivity of CuSeO3-x with electrons as the predominant charge carriers. This outcome is further corroborated by the negative Hall coefficients measured for the materials (Table S3)63,64. Table S4 lists the calculated electron densities for different CuSeO3-x in accordance with Eq. 2. It can be seen that the values are varied from 1.0 × 1021 to 2.3 × 1021 cm−3 as a function of Ov concentration, indeed capable of causing LSPRs in the NIR domain. These results, together with the observed linear correlation between the amounts of oxygen vacancies and free carriers in CuSeO3-x (Fig. 1h), substantiate that the strong absorption feature ranging from the red edge of visible to NIR observed for CuSeO3-x, should originate from LSPRs that are induced by the nonstoichiometric oxygen deficiency.
As illustrated in Fig. 2a, in the light of the different bonding patterns and bond lengths of Cu-O, the oxygen vacancies in CuSeO3-x could be categorized into four different types: Ov1-1 (1.92 Å and 2.00 Å, three-coordinate), Ov1-2 (2.00 Å and 2.3 Å, three-coordinate), Ov1-3 (2.00 Å and 2.00 Å, three-coordinate), and Ov2 (1.92 Å, two-coordinate). Synchrotron radiation-based X-ray absorption fine structure (XAFS) spectroscopy was employed to distinguish which type of oxygen vacancies dominates in CuSeO3-x. The obtained X-ray absorption near edge structure (XANES) spectra at Cu K edge are presented in Fig. 2b, where the similar intensities of white lines for CuSeO2.98 and CuO reveal that the Cu species in CuSeO2.98 are primarily in the form of Cu²+. Differently, the white line intensity for CuSeO2.82 is slightly lower than CuO, suggesting an enhanced electron density relative to Cu2+, in parallel with the findings acquired from XPS as above discussed (Fig. 1d). Figure 2c shows the Fourier transform (FT) of extended X-ray absorption fine structure (EXAFS) spectra for the samples, from which one can see that both CuSeO2.98 and CuSeO2.82 display only one major peak at 1.57 Å ascribed to Cu-O coordination. More importantly, the specific Cu-O bond length and Cu coordination numbers were extracted from the R space fitting curves of EXAFS (Fig. 2d and Table 1). It was uncovered that compared to CuSeO2.98 with less oxygen vacancies, CuSeO2.82 containing higher density of oxygen vacancies exhibit a marked decrease in the coordination number (from 3.3 to 2.8) exclusively at Cu-O with a bond length of 2.0 Å. Such information enabled us to unravel that the oxygen vacancies in CuSeO3-x are mainly located at the Ov1-3 site. The wavelet transform (WT) analysis of EXAFS signals was additionally performed, and similar conclusions are reached (Fig. 2e).
Fig. 2: Investigation on the local structure of Ov in CuSeO3-x.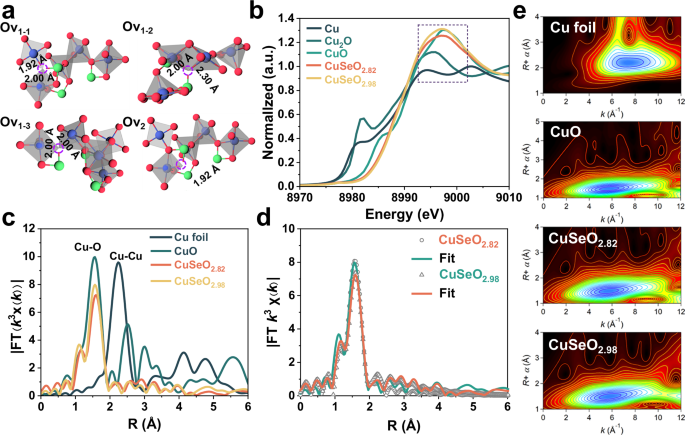
a Structural models showing different coordination environments for Ov in CuSeO3-x (Blue spheres represent Cu atoms; green spheres represent Se atoms; red spheres represent O atoms). b Normalized Cu K-edge XANES spectra, c k3-weighted FT-EXAFS spectra, d EXAFS fitting curves in R space and e WT-EXAFS contour maps. Source data for Fig. 2b–d are provided as a Source Data file.
Table 1 Fitting results of Cu K-edge EXAFS Data
It has been reported that some typical oxide plasmonic semiconductors like WO3-x often possess poor stability, as their oxygen vacancies are easily eliminated in O2-containing environment causing the loss of LSPRs effect50,65,66. Therefore, after resolving the origin of LSPRs and the local structure of oxygen vacancies in CuSeO3-x, we turned to evaluate the stability of Ov and the optical absorption properties for CuSeO3-x after light irradiation in air. In situ DRS and EPR measurements disclosed that after constant exposure to a Xe lamp (350 mW cm−2) for 24 h, no significant changes occurred to CuSeO2.82, except for a minor enhancement in the LSPRs band accompanied by a slight increment in the Ov concentration (Fig. S4). According to DFT calculations, the Ov formation energies in CuSeO3 and CuSeO2.82 are 1.49 and 1.89 eV, respectively, which are considerably lower than those in WO3 (4.45 eV) and other widely studied oxide photocatalysts (2.36 – 5.7 eV, details see Table S5), explaining the excellent Ov stability observed in our study. Furthermore, we found that by treating the as-prepared CuSeO2.82 with a mixture of H2/Ar gases at 300 oC, an extra amount of oxygen vacancies can be incorporated to give more pronounced LSPRs absorption with well retained crystallographic phases (Fig. S5, Table S1, the resulting material is denoted as CuSeO2.71-H2).
Photocatalytic methane oxidation with O2
The photocatalytic methane oxidation reaction was performed using molecular oxygen as the oxidant in a watersolution under ambient temperature (Fig. S6a). In specific, 20 mg of the CuSeO3-x photocatalysts were suspended in 20 mL of distilled water at 25 °C (temperature controlled by a circulating water bath) with CH4/O2 (v/v = 2/1, 0.1 MPa) as the feed gas under irradiation of a Xe lamp (350 mW cm−2). After 2 h of reaction, the bare CuSeO3-x material (in specific CuSeO2.90) showed near one unity selectivity in producing CH3OH (Fig. 3a), with no other products detected except for a negligible amount of CO2. To further improve the conversion efficiency, a series of cocatalyst particles were loaded including CuOx, FeOx, Ag, Au and Pt (Fig. S7). The results showed that Au/CuSeO2.90 afforded the highest CH3OH yield of 4026 μmol g−1 and a selectivity of 93.7% (Fig. 3a). It is noteworthy that for FeOx/CuSeO2.90 entirely composed of earth abundant elements, the CH3OH yield reached 1920 μmol g−1 (90.6% selectivity) comparable to those of Ag/CuSeO2.90 and Pt/CuSeO2.90.
Fig. 3: Photocatalytic performance in CH4 oxidation to CH3OH with O2.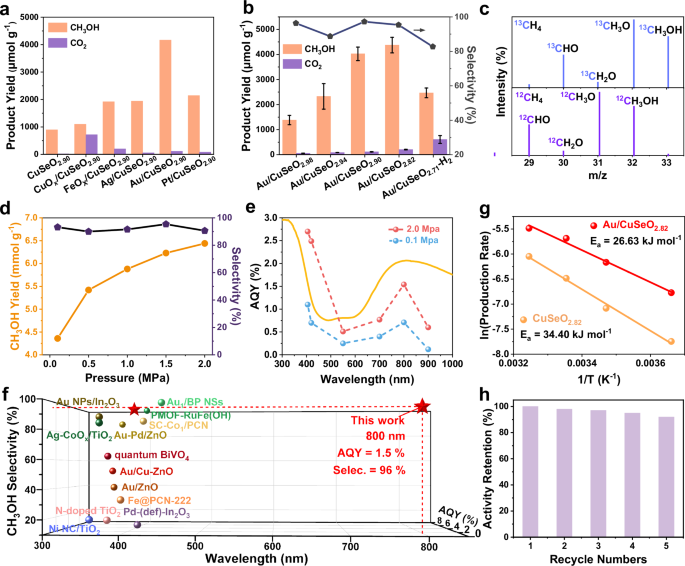
Influnences of (a) different cocatalysts and (b) different Ov concentraions. The error bars indicate standard deviation. c GC-MS results of the isotope labeling experiments for Au/CuSeO2.82 in the presence of 13CH4 or 12CH4. d Impacts caused by the pressure of reaction gas for Au/CuSeO2.82. e Wavelength-dependent AQY for Au/CuSeO2.82 under ambient and elevated pressures. f AQY and selectivity at different wavelengths for CH4-to-CH3OH conversion over Au/CuSeO2.82 in comparision with the state of the art (details see Table S8). g Arrhenius plots for Au/CuSeO2.82 and CuSeO2.82. h Recyclability measurement for Au/CuSeO2.82. Source data for a–e, g, h are provided as a Source Data file.
After screening the cocatalysts, the influences of Ov concentration on photocatalytic performance were assayed. According to Fig. 3b, the optimal concentration of oxygen vacancies was given by CuSeO2.82. With Au cocatalsyts (Au particle size centered at 10.2 nm, Fig. S8), this sample exhibited a CH3OH yield of 4373 μmol g−1 with 93.4% selectivity. Further increasing the Ov concentration led to a depressed CH3OH yield, as demonstrated by Au/CuSeO2.71-H2 (likely containing an excessive amount of Ov thereby enhancing the probability of charge recombination, Fig. S9). Besides, the impacts of molar ratio between CH4 and O2 as well as the amount of H2O were investigated (Fig. S10). The results showed that under optimal condition (CH4/O2 = 2/1, 0.1 MPa, 40 mL of H2O) the CH3OH yield over Au/CuSeO2.82 can be further increased to 4780 μmol g−1 (corresponding to an averaged formation rate of 2.4 mmol g−1 h−1), while the selectivity was nearly unchanged at 95.3% with CH3OH as the sole liquid oxygenate product. To ensure that the detected CH3OH was generated from CH4 oxidation, control experiments were carried out, where no products were generated with the absence of photocatalyst, light, CH4 or O2 (Fig. S13). Furthermore, 13C isotope labeling experiments using 13CH4 as reactant additionally confirmed the carbon source for CH3OH collected after the photocatalytic reaction (Fig. 3c). Besides, carbon balance for the reaction system was determined to be 93% (Table S6), strongly confirming the absence of other side products.
When raising the pressure of CH4 to enhance its solubility in water, the CH3OH yield over Au/CuSeO2.82 can be further improved to 6440 μmol g−1 under 2.0 MPa with little perturbed selectivity (Fig. 3d, Fig. S6b). Regarding this sample, the AQY for CH3OH generation was measured at different excitation wavelengths under atmospheric (0.1 MPa) or elevated pressure (2.0 MPa) with temperature fixed at 25 °C (Fig. S14, Table S7). As displayed in Fig. 3e, under both conditions the action spectra for CH4-to-CH3OH conversion basically follows the optical absorption of the material across the UV-Vis-NIR range. At ambient pressure, the AQY was determined to be 1.1% (92.7% selectivity) at 405 nm, and this value was improved to 2.7% (94.6% selectivity) when pressure was increased to 2.0 MPa. Remarkably, the results also showed that with light irradiation at longer wavelengths, for example λ = 800 nm, the AQY was of 0.7% (93.4% selectivity) at 0.1 MPa while reached 1.5% (96.2% selectivity) at 2.0 MPa. Figure 3f summarizes the results of AQY and selectivity with respect to CH3OH production reported for the state-of-the-art photocatalysts enabling CH4 partial oxidation by O2 (25 °C, 0.8–3.0 MPa, details see Table S8)10,19,20,21,22,23,24,25,26,67,68,69,70. It is worth underlining that although significant advances have been achieved, effective CH4 conversion into C1 oxygenates driven by photons with wavelengths exceeding 450 nm is still very challenging and has rarely been realized in previous studies9,10,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,67,68,69,70,71,72,73. Here, our results present a stride in this scope, where NIR light can be harnessed to activate CH4 into CH3OH with exceptional activity and selectivity. This gives an unambiguous demonstration that the long-wavelength photons are well capable of overcoming the energy barrier in CH4 oxidation under mild conditions, mapping an attractive route for improving the utilization efficiency of solar energy to transform CH4 into value-added chemicals.
The apparent activation energy for photocatalytic CH4-to-CH3OH conversion was then estimated by fitting the measured temperature-dependent reaction rates with Arrhenius equation, in the temperature range of 0 to 35 °C. The Ea values were determined to be 26.6 kJ mol−1 (0.28 eV) and 34.4 kJ mol−1 (0.36 eV) for Au/CuSeO2.82 and CuSeO2.82, respectively, under illumination (Fig. 3g, Ea in the dark cannot be acquired due to the lack of activity). Such low Ea results suggest that the plasmon-mediated hot carriers might greatly assist the activation process of CH4, as it has been demonstrated that illumination of a plasmonic photocatalyst can dramatically reduce the Ea relative to the thermal case34,35,36,37. In view of the much higher AQY at 800 nm than that at 550 nm (near the LSPR band of Au nanoparticle) for Au/CuSeO2.82 (Fig. 3e), as well as the poor activity observed for the bare Au nanoparticles (CH3OH formation rate of 24 μmol g−1 h−1 under 2.0 MPa, details see Fig. S15), we reasonably assume that the LSPRs of CuSeO2.82 should make a major contribution in modifying the reaction kinetics. In addition to its excellent catalytic performance, Au/CuSeO2.82 also demonstrated high recyclability, retaining 92% of its initial catalytic activity after five consecutive reaction cycles (10 h total irradiation, 2.0 MPa, Fig. 3h). The samples collected after photocatalytic reaction exhibited good structural and compositional stability as indicated by combined XRD, XPS and EPR characterizations (Fig. S16).
Mechanism investigation
With regard to plasmonic photocatalysis, the hot carrier-promoted chemical conversion might be entangled by thermal effects. To distinguish the thermal and nonthermal contributions, the surface temperature of the photocatalyst powder was measured first using a thermal imaging camera (Fig. S17a). It was found that the photothermal heating of Au/CuSeO2.82 was not very significant, with the surface temperature reaching a plateau at 88 °C after light illumination (350 mW cm−2) for 300 s. Moreover, when dispersing Au/CuSeO2.82 into the aqueous reaction solution, the temperature of the system can only be raised to 42 °C by light illumination (Fig. S17b). At this reaction temperature, the yield of CH3OH was evidently depressed whilst the production of HCHO and CO2 was promoted in comparison to those at 25 °C (Fig. S18). These outcomes manifest that in our experiments (reaction temperature controlled at 25 °C by circulating water bath) the impacts of photothermal heating on catalytic performance were negligible.
Next, the influences of oxygen vacancies on photogenerated charge separation properties were systematically explored. In Fig. 4a, upon illumination the open-circuit potential (OCP) of the CuSeO3-x samples all presented a prompt negative shift, in agreement with the n-type conductivity as above discussed61,74. The saturated OCP response denotes the steady state density of light-excited carriers as a result of the competition between their production and recombination rates. One can see that this value increased in the order of CuSeO2.98 2.94 2.90 2.82, clearly indicating that the increase in Ov concentration in these samples can lower the rates of photogenerated carrier recombination. The photocurrent as well as electrochemical impedance spectroscopy (EIS) measurements also supported that the optimal Ov density in CuSeO3-x are propitious for promoting the charge separation and transfer efficiencies (Fig. S9).
Fig. 4: Identification of the active sites and reactive species.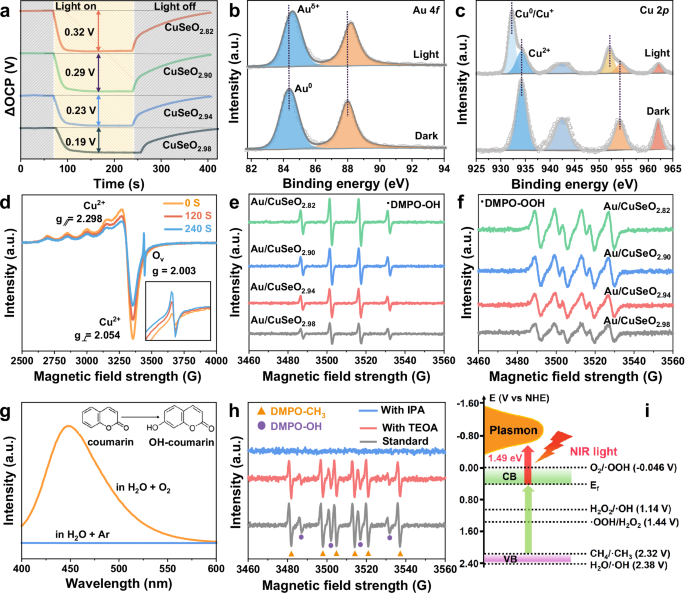
a OCP response under illumination and in the dark. In situ high-resolution (b) Au 4 f and (c) Cu 2p XPS spectra, as well as (d) in situ EPR spectra for Au/CuSeO2.82 in the dark and under light irradiation. In situ EPR spectra of (e) DMPO-OH and (f) DMPO-OOH over different photocatalysts under light illumination. g Fluorescence spectra of the coumarin aqueous solution dispersed with Au/CuSeO2.82 under light irradiation in O2 or Ar atmosphere. Insets: the detection mechanism of •OH by coumarin. h In situ EPR spectra of DMPO-OH and DMPO-CH3 over Au/CuSeO2.82 under light illumination in the presence of sacrificial agent or not. TEOA is used for consuming photogenerated holes, and IPA is used for consuming photogenerated holes and •OH radicals simultaneously. i Illustration of the band structure determined for Au/CuSeO2.82 and the redox potentials for different radicals. Source data for a–h are provided as a Source Data file.
In situ high resolution XPS was exploited to determine the charge transfer direction and chemical nature on the surface of Au/CuSeO2.82 under light illumination. As shown in Fig. 4b, the binding energy of Au 4 f shifted positively from 84.3 to 84.6 eV upon irradiation, disclosing that the Au cocatalysts were able to accumulate photogenerated holes. On the contrary, the Cu 2p3/2 and Cu 2p1/2 signals presented notable negative shifts (to 932.2 and 952.1 eV, respectively) after illumination, along with evident decrease in the intensity of satellite peaks (Fig. 4c). These observations can be rationalized by the reduction of Cu2+ under light, which disclosed that the Cu2+ ions in CuSeO3-x played the role of electron acceptors. A similar conclusion can be derived from the in situ EPR spectra exhibited in Fig. 4d. The EPR signals at g⊥= 2.054 and g∥= 2.298 attributed to Cu2+ gradually decreased as a function of illumination time, indicative of the conversion of Cu2+ into EPR-silent species20. Such process accordingly enhanced the amount of oxygen vacancies nearby (g = 2.003) as can be discerned from the inset of Fig. 4d.
Moreover, by means of in situ EPR using 5,5-dimethyl-1-pyrroline-N-oxide (DMPO) as radical-trapping agent, we uncovered that reactive •OH and •OOH radicals were generated over Au/CuSeO3-x photocatalysts in the presence of O2 and H2O under irradiation20,24,33. Figure 4e, f clearly show that the signal intensities for both of these two species were positively correlated with the density of oxygen vacancies in the materials, with Au/CuSeO2.82 offering the highest capability for delivering •OH and •OOH. On account of these results as well as the trapping of electrons at the Cu2+ sites, we propose that the Cu2+ and neighboring oxygen vacancy (CuII-Ov) may cooperatively compose an active center for collecting photoexcited electrons and reducing O2 into •OH and •OOH. However, it should be taken into mind that in an environment containing O2 and H2O, water oxidation provides an alternative path to generation of •OH and •OOH radicals. To identify the origin of these radicals in our reaction, fluorescence measurements with coumarin as probe molecules were performed and the results are given in Fig. 4g22,26. One can see that for Au/CuSeO2.82 dispersed in H2O under Ar atmosphere, no fluorescence signal of 7-hydroxycoumain (OH-coumarin) can be detected after light illumination. In contrast, the introduction of O2 into the system led to a pronounced signal attributed to OH-coumarin. The sharp discrepancy in the presence of O2 or not strongly substantiated that in our experiments the •OH (and thus •OOH) radicals were dominantly produced via O2 reduction other than H2O oxidation. This conclusion was further corroborated by in situ EPR measurements exhibited in Fig. S19, where substitution of O2 with Ar in the aqueous suspension of Au/CuSeO2.82 resulted in disappearance of both DMPO-OH and DMPO-OOH signals under light irradiation. Aside from these reactive oxygen species, the generation of H2O2 during O2 reduction process was also confirmed by the iodometric titration method (details see Fig. S20). Combining the information gleaned above, we propose that the photoreduction of O2 over Au/CuSeO2.82 should go through a stepwise process via O2 → •OOH → H2O2 → •OH25,28.
As regards the oxidation of CH4, we found that in the photocatalytic reaction system, very strong EPR signals corresponding to CH3• appeared along with •OH upon light illumination, and both signals intensified with increasing irradiation time (Fig. S21)68. More significantly, Fig. 4h shows that when introducing triethanolamine (TEOA) into the reaction system to sacrifice photogenerated holes, the intensity of the EPR signals arising from CH3• was slightly decreased. However, the addition of isopropyl alcohol (IPA), which is able to consume the •OH radicals and holes simultaneously, almost completely ceased the generation of CH3•. According to these results, we inferred that in our case the in situ generated •OH should function as the primary reactive species that promote CH4 activation.
On the basis of Tauc plot, valence band XPS (VB-XPS) spectrum and Mott-Schottky measurements (Fig. S22), the conduction band (CB) and valence band (VB) of Au/CuSeO2.82 were evaluated at 0.34 and 2.35 V vs NHE, respectively (Fig. 4i). Considering the oxidation potential of •OH/H2O at 2.38 V vs NHE, the VB position of the material cannot provide a large driving force for H2O oxidation to •OH. Similarly, the activation of CH4 straightforwardly by photogenerated holes in VB did not possess much advantage in thermodynamics (•CH3/CH4 = 2.32 V vs NHE). In sharp contrast, the surface plasmons of excited Au/CuSeO2.82 with energies 1.49 eV (corresponding to the resonant excitation at 830 nm) above the Fermi level, can afford substantial driving force for reducing O2 and the involved intermediates as illustrated in Fig. 4i.
To gain deeper insights into the mechanisms underlying O2 activation and conversion during photocatalytic CH4 oxidation process, DFT calculations were implemented on CuSeO3 (111) facet with or without oxygen vacancies (Fig. 5a–e). First, the O2 adsorption behavior was examined on the two different surfaces. The results unraveled that the incorporation of oxygen vacancies can markedly promote O2 adsorption, in terms of the enhanced adsorption energy (−0.26 vs −0.11 eV), shortened spatial distance (2.00 vs 2.57 Å) and increased O-O bond length (1.27 vs 1.24 Å, Fig. 5a and Fig. S23). Noteworthily, the O2 molecules are preferred to be adsorbed at the Cu atom directly adjoining the oxygen vacancy, supporting our assumption that the CuII-OV associative sites are responsible for the activation of O2 molecules. On the contrary, the presence of oxygen vacancies brings about negligible impacts on the adsorption capacity for CH4 (Fig. S24). Thereafter, the thermodynamics associated with O2 reduction during the reaction via proton-coupled electron transfer (PCET) pathway was estimated. As displayed in Fig. 5e, after the adsorption of O2, both CuSeO3 and CuSeO3-x undergo similar conversions through O2 → *OOH → *H2O2 → *OH, in good agreement with our experimental observations discussed earlier. A distinct advantage is that these entire processes are thermodynamically spontaneous with each steps exhibiting a notable decrease in Gibbs free energy, indicative of the exceptional capability for activating O2 into reactive *OH species. Apparently, compared to the Ov-free CuSeO3, CuSeO3-x exhibits even larger driving force for the sequential transformation in view of the steeper energy declines (for example, −0.7 vs −1.47 eV in *H2O2 dissociation step). From structural analysis in Fig. 5a–d, this can be attributed to the CuII-Ov sites which function as the reaction center to stabilize lower energy states. Such kinetically favorable production of *OH is strongly desired for CH4 activation and the subsequent coupling reaction to yield CH3OH in high selectivity. Beyond efficient O2 to *OH conversion, CuSeO3-x also demonstrates its superiority in suppressing over-oxidation according to DFT calculations. As manifested in Fig. S25, the substantial energy difference between *CH3OH desorption and further oxidation (0.58 vs 1.54 eV) makes the generation of higher oxidation products evidently thermodynamically unfavorable.
Fig. 5: Evaluation of reaction pathway.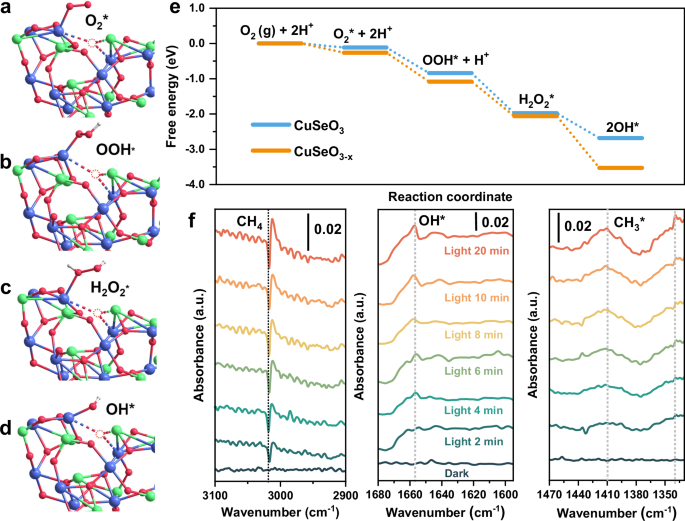
a–d Illustration of oxygen adsorption and reduction configurations at the CuII-Ov active sites in CuSeO3-x (Blue spheres represent Cu atoms; green spheres represent Se atoms; red spheres represent O atoms; white spheres represent H atoms). All geometry-optimized computational models and corresponding atomic coordinates originate from the file Supplementary Data 1. e Comparison of the calculated energy profiles on the surface of CuSeO3-x and CuSeO3 for O2 adsorption and activiation. f In situ DRIFTS spectra for Au/CuSeO2.82 during the photocatalytic methane oxidation reaction. Source data for e, f are provided as a Source Data file.
Furthermore, in situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) was carried out over Au/CuSeO2.82 in the presence of CH4, O2 and water vapor (Fig. 5f). With constant light irradiation, an evident decrease at 3018 cm−1 ascribed to the antisymmetric stretching of CH4 was observed75,76,77, confirming the effective consumption of CH4 over the photocatalyst. More importantly, several new bands emerged and grew continuously during the irradiation process. In specific, the peak at 1654 cm−1 was probably stemmed from the bending signal of OH*27,71,76,77, while those at 1412 and 1339 cm−1 could be assigned to the scissoring and symmetric rocking vibrations of CH3*, respectively27,73,75,77. These observations are well aligned with the overall picture proposed for photocatalytic CH4 oxidation on surface of Au/CuSeO3-x as illustrated in Fig. S26: under light irradiation, LSPRs of CuSeO3-x are excited. The generated hot electrons are trapped by CuII-Ov sites to reduce O2 into •OH via multiple PCET steps. In the meantime, the photogenerated holes accumulate on Au cocatalysts, generating positively charged Auδ+ sites where the CH4 molecules could be activated into CH3• radicals by in situ produced •OH17,78. The combination of CH3• and •OH then yields CH3OH molecules, which can be easily dissolved in water avoiding overoxidation on the photocatalyst surface.
In summary, a new type of plasmonic semiconductor has been created by manipulating the oxygen vacancies in copper oxyselenide. The resultant CuSeO3-x can efficiently capture NIR photons to accomplish CH4-to-CH3OH transformation at ambient temperature with O2 as the oxidant. Such fascinating performance is strongly associated with the CuII-Ov sites on surface of CuSeO3-x, which enable a kinetically feasible route to reduce O2 into •OH radicals, markedly lowering the energetic barrier for CH4 activation and oxidation. This work showcased the first demonstration that low-energy NIR photons can be utilized as the sole stimulus to drive CH4-to-CH3OH conversion, which conventionally suffers from excessive energy depletion due to high activation energies. In addition, our study opened up new possibilities in expanding the currently limited pool of plasmonic semiconductors, to develop superior photocatalysts in response to the worldwide increasing demand of sustainable chemical feedstock and fuels.