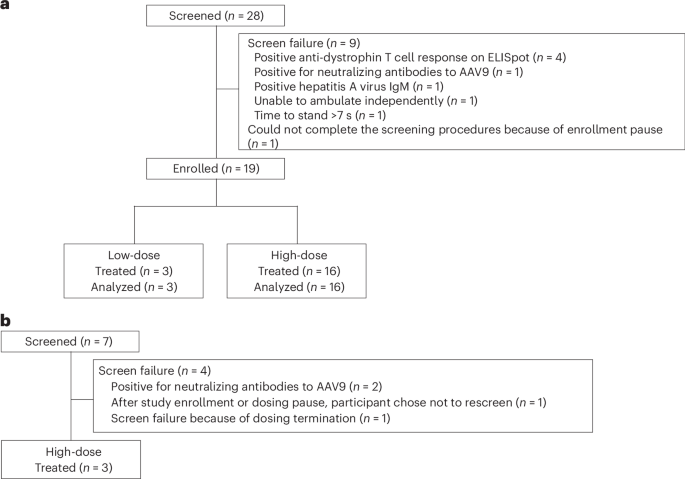
Study design and oversight
This ongoing, nonrandomized, open-label, ascending-dose, phase 1b study (ClinicalTrials.gov registration no. NCT03362502) was initiated in January 2018 at three sites in the United States (Supplementary Fig. 3 and detailed in the Supplementary Protocol) and was conducted in compliance with the ethical principles of the Declaration of Helsinki 2013 and all International Conference on Harmonisation Good Clinical Practice Guidelines. The protocol was approved by the relevant institutional review board (IRB) or independent ethics committee at each study site (University of Utah IRB, Duke University Health System and UCLA Medical IRB). All participants (or parent or legal guardian) provided written informed consent. Data collection occurred at each study site (study start date 23 January 2018; primary completion date 28 March 2022, ClinicalTrials.gov registration no. NCT03362502). Participants from three sites in the United States were enrolled in the trial.
Key inclusion and exclusion criteria
Personally signed and dated informed consent or assent (where appropriate) documents were obtained, indicating that the participant or a legally acceptable representative, parent(s) or legal guardian was informed of all pertinent aspects of the study and were willing and able to comply with scheduled visits, treatment plans, laboratory tests and all other required study procedures. Participants were not compensated.
Eligible ambulatory males were aged 4–12 years (inclusive), had a body weight of 15–50 kg (inclusive), had a diagnosis of DMD (confirmed by medical history and genetic testing before screening), were able to tolerate magnetic resonance imaging and muscle biopsies under anesthesia without sedation, with no contraindications to these procedures. Participants were receiving daily glucocorticoids for 6 or more months (stable regimen for 3 or more months before baseline). Ambulatory status was defined as able to walk 10 m unassisted and rise from the floor within 7 s.
Eligible nonambulatory male participants were of any age (ambulation before the participant’s 17th birthday), had a body weight of 75 kg or lower, had a diagnosis of DMD (confirmed by medical history and genetic testing before screening) and were receiving daily glucocorticoids for 6 or more months (stable regimen for 3 or more months before baseline). Nonambulatory status was defined as unable to walk 10 m unassisted and rise from the floor within 7 s, with adequate respiratory function (percentage predicted forced vital capacity (FVC) greater than 40%), adequate upper-limb function (v.2.0), and an entry item A score of 3 or more.
All participants with neutralizing antibodies against the AAV9 capsid at screening were excluded. Additional exclusion criteria included receipt of a live attenuated vaccination (within 3 months) or exposure to an influenza vaccination or systemic antiviral or interferon therapy (within 30 days) before receiving the study treatment; current exposure to systemic immunosuppressant agents other than glucocorticoids; genetic abnormalities in the dystrophin gene as confirmed by the investigator based on the review of DMD genetic testing, that is, any mutation (deletion, duplication, insertion or point mutation) affecting any exon between exons 9 and 13 inclusive; or deletion affecting both exons 29 and 30; prior gene therapy treatment (any therapy introducing exogenous DNA or intended to permanently alter endogenous DNA); gene therapy (other than the study drug); exposure within 6 months before screening (visit 1) to any treatment designed to increase dystrophin expression (including, but not limited to exon-skipping agents and nonsense read-through). These treatments were also prohibited during the period between screening (visit 1) and day 1 (visit 3) and for the first year of the study; participation in other studies involving investigational drug(s) within 30 days or five half-lives, whichever was longer, before the baseline visit; known hypersensitivity to any of the components of the study drug or solution for infusion, such as hypersensitivity to albumin or a diagnosis of (or symptoms suggestive of) hereditary fructose intolerance; presence or history of other musculoskeletal or neurological diseases in addition to DMD; evidence or history of clinically significant hematological, renal, endocrine, pulmonary, gastrointestinal, cardiovascular, hepatic, cancer, autoimmune or allergic disease (including drug allergies, but excluding untreated, asymptomatic, seasonal allergies at the time of dosing); LVEF lower than 55% (LVEF lower than 35% for nonambulatory participants), specified abnormalities in hematology or chemistry tests (absolute neutrophil count less than 1,000 cells per mm3; cystatin C greater than 1.2 times ULN; platelets lower than 150 × 103 µl−1); positive hepatitis A virus IgM; and hepatitis B surface antigen or hepatitis C antibody. Additional exclusion criteria were serum markers indicating possible autoimmune-mediated hepatitis: antinuclear antibody titer greater than 1:160; total IgG more than two times the ULN; markers of hepatic inflammation or overt or occult cirrhosis as evidenced by one or more of the following: total bilirubin more than 1.5 times the ULN and direct bilirubin ≥0.5 mg dl−1; GGT test more than 1.5 times the ULN; prothrombin greater than the ULN; prolonged international normalized ratio greater than the ULN; and GLDH more than two times the ULN. Participants with clinically significant infection within 30 days before study drug administration, and acute or chronic medical or psychiatric conditions, including recent (within the past year) or active suicidal ideation or behavior (within the past 6 months), or laboratory abnormality that may increase the risk associated with study participation or investigational product administration or may interfere with the interpretation of the study results were also excluded. In addition, nonambulatory participants were excluded if there was any injury that may have impacted functional testing. Previous injuries must have fully healed before consenting. For ambulatory participants, prior lower-limb fractures must have fully healed and at least 3 months from the injury date.
Study treatment
All ambulatory participants received a single intravenous infusion of low-dose (1 × 1014 vector genomes per kilogram of body weight; 1 × 1014 vg kg−1) or high-dose (3 × 1014 vg kg−1) fordadistrogene movaparvovec, according to dose escalation rules (Methods). Information on the vector and transgene has been described previously25,36. All nonambulatory participants received a single intravenous infusion of high-dose fordadistrogene movaparvovec. Vector genome concentration was initially determined via an inverted terminal repeat assay and later transitioned to a transgene-based quantitative PCR assay. For the high dose, 3 × 1014 vg kg−1 using the inverted terminal repeat assay is approximately equivalent to 2 × 1014 vg kg−1 using the transgene-based assay.
To mitigate the risk of immune responses, participants were administered a prespecified glucocorticoid regimen during the first 3 months (Methods). After 3 months, participants resumed their baseline glucocorticoid regimen and continued this regimen throughout the first year of follow-up, except for adjustments due to AEs or changes in body weight. Details on the use of concomitant medication, including rescue medication to manage immune responses, are provided in the Methods.
Endpoints and assessments
The primary endpoints were dose-limiting AEs, and safety and tolerability through 1 year after treatment based on the incidence, severity and causal relationship of TEAEs. These were coded using the Medical Dictionary for Regulatory Activities v.24.0, with severity and relationship to treatment determined by the site investigators. Other safety assessments included the incidence and magnitude of abnormal findings from clinical laboratory tests, physical and neurological examinations, electrocardiograms, LVEF (assessed using magnetic resonance imaging or echocardiogram) and C-SSRS, which were conducted throughout the study through to week 52 (Supplementary Fig. 3).
The secondary endpoint was expression of mini-dystrophin in biceps brachii muscle biopsy samples. Total dystrophin concentration LC–MS and the proportion of mini-dystrophin-positive fibers (assessed using automated image analysis of immunofluorescence) were determined at baseline, 2 months and 1 year after treatment (Methods). The LC–MS assay used for the assessment recognized both full-length dystrophin and mini-dystrophin, while the immunofluorescence assay used a mini-dystrophin-specific antibody; thus, it detects the mini-dystrophin transgene only. The term ‘dystrophin’ is used to describe both forms of the protein.
Assessment of dystrophin and mini-dystrophin using LC–MS and immunofluorescence
The LC–MS assay used for the quantification of dystrophin and transgene-expressed mini-dystrophin consisted of the following steps: (1) skeletal muscle homogenization in a sodium dodecyl sulfate containing lysis buffer; (2) protein precipitation using acetonitrile; (3) digestion with trypsin to yield the surrogate peptides used for quantification: (4) enrichment of these peptides using antipeptide antibodies; and nanoflow LC coupled to a triple quadrupole mass spectrometer operating in multiple reaction monitoring mode. Stable isotope labeling by amino acids in cell culture mini-dystrophin was used as an internal standard and calibration of the assay was achieved using a mini-dystrophin recombinant protein. The assay was validated as described previously14.
The mini-dystrophin/laminin wet chemistry, and image analysis, assays were fully validated before use in this study (Flagship Biosciences). Briefly, slides were fluorescently labeled with mini-dystrophin/laminin and scanned on a 3DHISTECH Pannoramic SCAN II fluorescence scanner in the green and red channels at ×20. Manual annotations were applied across the entire tissue to identify regions of analysis for each specimen to be scored by image analysis with inclusion annotations to capture relevant target tissue (ice, muscle fibers) and exclusion annotations to remove regions on the slide image containing either unanalyzable tissue (for example, necrosis, folding, dust, crush artifacts, other tissue-specific artifacts) or analyzable tissue that was not of interest. The Flagship Biosciences proprietary image analysis algorithm was used for classification and quantification of the staining intensity of the background-corrected red channel mini-dystrophin (representing mini-dystrophin) in each fiber identified by the developed algorithm. The algorithm collected various endpoint data on each fiber in the biopsy. The primary data reported for this study were derived from: (1) the total number of fibers analyzed on the slide; (2) the number of mini-dystrophin-positive fibers; and (3) the mean stain density (MSD) of mini-dystrophin labeling for all fibers. A global threshold for calculating the percentage of mini-dystrophin-positive fibers was established based on all baseline samples from the 19 ambulatory participants. The quantification of the immunofluorescence intensity level (MSD) associated with the individual muscle fibers from these 19 participants was used to calculate the 99th percentile of the overall MSD distribution. The corresponding MSD threshold value was determined. Any muscle fiber with an MSD value above this calculated threshold was considered as a positive fiber. The antibodies, controls and concentrations used for immunofluorescence are shown in Supplementary Table 5. Two sections from each muscle biopsy were analyzed for immunofluorescence; the average results from the two sections were reported per visit per participant.
Prespecified exploratory functional endpoints included change from baseline to 1 year in the NSAA total score34, time to rise from floor, time to climb four stairs, 6-min walk distance, time to walk/run 10 m (10-m w/r), performance of the upper limb 2.0 (ref. 37) and percentage predicted FVC (%pFVC). Refinement of two exploratory endpoints were made: the number of NSAA items (that is, skills) gained (among participants with at least one baseline individual item score of zero) and the number of skills maintained or improved (among all participants). Patient-reported and parent-reported assessments (as measured using the Pediatric Outcomes Data Collection Instrument assessment) were also included and have been described separately38.
Major protocol amendments relevant to the ambulatory population reported in this study
The protocol and amendments were approved by the IRB and independent ethics committee for each site (University of Utah IRB, Duke University Health System and UCLA Medical IRB).
Amendment 1 detailed the prohibition of influenza vaccine within 30 days before and 3 months after the administration of fordadistrogene movaparvovec, and allowed for flexibility of weight-based calculations for required glucocorticoids to align with standard of care.
Amendment 2 detailed the requirement for observation at the site after administration of fordadistrogene movaparvovec until any observed events had been resolved residing near the investigational site for at least 1 week for ease of follow-up and allowed for medications to treat or prevent side effects, such as antiemetics or antihistamines for nausea and vomiting.
Amendment 3 detailed the requirement to reside near the investigational site for at least 2 weeks, and an additional visit on days 10 and 21 (added to days 4, 7 and 14) for clinical and laboratory assessments. This amendment also detailed the addition of triplicate electrocardiogram at weeks 2 and 4, safety labs added through week 8 to monitor for activation of the complement system and associated clinical findings (including blood smear for schistocytes, haptoglobin and exploratory immune biomarkers) and requirement for eculizumab to be accessible by the investigational sites for use if complement activation was observed. In addition, Amendment 3 detailed the requirement for meningococcal vaccination (at least 2 weeks before dosing) or, if vaccination was contraindicated, prophylactic antibiotics for meningococcus if eculizumab was administered, and allowed for flexibility in timing of after treatment muscle biopsies (that is, at either 2 or 3 months and either 6 or 12 months; note that a single participant underwent a posttreatment biopsy at 3 instead of 2 months; all planned second posttreatment biopsies were performed within 1 month of the 12-month visit).
Amendment 4 detailed the number of enrolled individuals to be increased from 12 to 24, removed the exclusion criterion pertaining to a preexisting T cell response on ELISpot assay, the addition of possible local laboratory samples to be collected and tested on study days 7–10 and extension of complement biomarker collection through week 8. This amendment also detailed the assessment of drug concentrations of fordadistrogene movaparvovec changed to the transgene method, per regulatory request, and increased flexibility in the dosage of protocol-required glucocorticoids just before (that is, intravenous methylprednisolone) and immediately after (that is, oral prednisone) administration of fordadistrogene movaparvovec.
Amendment 5 detailed the requirement for local laboratory testing on days 5–9, and removed the anti-smooth muscle antibody assay and anti-liver kidney microsomal antibody type 1 laboratory tests for autoimmune-mediated hepatitis as exclusion criteria, given the lack of observed hepatic injury associated with fordadistrogene movaparvovec. This amendment also detailed the increased dose of intravenous methylprednisolone from ≥1 to ≥2 mg kg−1, increased the dose of oral prednisone/prednisolone from ≥1 mg kg day−1 to ≥2 mg kg day−1 for the first 2 weeks after administration of fordadistrogene movaparvovec and included all types of vaccinations (such as those potentially available for severe acute respiratory syndrome coronavirus 2) that are prohibited 30 days before and 3 months after the administration of fordadistrogene movaparvovec.
Staggered dosing design
To mitigate unanticipated risks to participant safety, enrollment was staggered within and between the fordadistrogene movaparvovec (PF-06939926) low-dose and high-dose groups and included a formal review by an external data monitoring committee (EDMC) before dose progression and in the event of any possible safety signals. In each of the dose-level groups, the dosing interval between the first and second participants was at least 6 weeks. If no safety concerns were identified 3 weeks after the second participant was infused, dosing proceeded at ≥3-week intervals. Similarly, if no stopping or pause criteria were met after dosing six consecutive participants at the high-dose level, dosing proceeded at that same dose level at ≥1-week intervals.
For both dose groups, enrollment was paused to accommodate EDMC review if any of the following occurred: any potentially treatment-related serious AE; similar clinically significant safety findings in 50% or more participants at a given dose level (indicating dose-limiting intolerance); repeated alkaline phosphatase or total bilirubin more than two times the ULN; repeated GLDH more than 2.5 times the ULN; a clinical diagnosis of myositis or myocarditis; other findings that, at the discretion of the sponsor study team, investigator or EDMC, indicated that dose escalation should be halted or that dose de-escalation would be appropriate.
In addition to the review of safety data, the 2-month muscle biopsy findings of the three enrolled participants in the low-dose group were reviewed by the EDMC before progressing to dosing of the high-dose group.
Administration of fordadistrogene movaparvovec
Fordadistrogene movaparvovec was supplied as a sterile frozen solution (5-ml volume) in a clear, single-use vial. The solution was thawed, diluted (in a 250–500-ml solution containing 1.25% serum albumin) and administered intravenously over approximately 2–4 h to participants by qualified site staff (for example, physician, nurse or pharmacist). Participants were monitored throughout infusion and for at least 24 h after infusion completion.
Glucocorticoid regimen
To mitigate the risk of an immune response and drug-induced liver injury, each participant was administered a prespecified glucocorticoid regimen over the first 3 months of the study. The regimen was revised in the protocol amendments (described above) and is summarized across all participants, including as a single intravenous dose of methylprednisolone (1–2 mg kg−1) 1–4 h before receiving the fordadistrogene movaparvovec infusion and daily oral prednisone or prednisolone after treatment (1–2 mg kg−1 for the first 2–4 weeks, tapering to 1.0 mg kg−1 through the end of the first month, 0.75 mg kg−1 for the second month and the higher dose of 0.50 mg kg−1 or the pre-study dose for the third month). After 3 months, participants resumed their baseline glucocorticoid regimen and remained on this regimen throughout the first year of follow-up, except for adjustments due to side effects or changes in body weight.
Use of concomitant medications
Participants could receive angiotensin-converting enzyme inhibitors, beta-blockers, angiotensin II receptor blockers and aldosterone blocker or thiazide diuretics if initiated at least 3 months before screening and stable dosing was planned during the study. Antiemetics were allowed in the first 7–10 days after dosing of fordadistrogene movaparvovec. Other permitted medications included dietary supplements and bisphosphonates.
Medications prohibited throughout the study included immunosuppressant agents (other than glucocorticoids) unless administered in response to immunological reaction, systemic antiviral or interferon therapy unless administered to treat an acute viral infection, other investigational therapies, other gene therapy agents and sedatives for imaging assessments. Treatments aiming to increase dystrophin expression were prohibited from screening through 1 year after treatment. Receipt of vaccinations (including influenza, live attenuated vaccines, mRNA-based or DNA-based vaccines, or nonreplicating viral vaccines) was prohibited from screening through 3 months after treatment.
Rescue medication
In the event of an infusion-site reaction during the infusion period, administration of treatment could be paused and supportive therapy could be provided according to standard of care (for example, antihistamine therapy). In the event of elevated liver enzymes during the study, glucocorticoid doses could be increased and immunomodulatory treatment could be added as needed after consultation with immunopharmacological or hepatic experts. In response to an AE due to apparent activation of the complement system (for example, abnormality in complement biomarkers, blood smear indicative of hemolysis or more than two consecutive reductions in platelet count), participants could receive at least one dose of the complement inhibitor eculizumab. As eculizumab increases the risk of contracting meningococcal infections, all previously unvaccinated participants were required to receive at least one dose of a meningococcal vaccination at least 30 days before receiving fordadistrogene movaparvovec. In case of vaccine allergy, prophylactic antibiotics for meningococcus were provided along with eculizumab.
Derivation and analysis of the external control cohort
The responsibility of the internal independent team was to (1) develop the key criteria for the selection of appropriate external control participants from the identified data sources; (2) identify the external control data, including detailed assessment of sources of potential bias and confounders; and (3) identify key covariates to include in the PS analyses.
Evaluation of comparability between the external control data sources and the current study were based on Pocock’s criteria of exchangeability4. Based on the independent team’s evaluation, the following criteria (evaluated at baseline) were used for the selection of the external control cohort: (1) aged 4–12 years at screening, inclusive; (2) an ability to ambulate independently at screening (or baseline if screening was missing), defined as a score of 1 or 2 on NSAA item 17 (10-m run/walk), or a score of 0 (not missing data) on NSAA item 17, or a score of 0 (missing data) for NSAA item 17 with a score of 1 or 2 for NSAA item 2 (walk); (3) an ability to rise from the floor within 7 s (as part of the NSAA) at screening (or baseline if screening was missing); (4) LVEF greater than 55% at screening (or at baseline if screening was missing); and (5) nonmissing NSAA total score at baseline (or screening if baseline was missing).
PS analysis was used to adjust for any remaining potential imbalance in baseline demographic and clinical characteristics between the treatment group and the external control cohort. The PS is the probability of treatment assignment conditional on observed baseline characteristics. It allows the analysis of an observational (nonrandomized) study in a way that mimics some of the particular characteristics of a randomized controlled trial5. PS were estimated using a multiple logistic regression model that incorporated potential treatment predictors as independent variables and treatment group as a dependent variable (fordadistrogene movaparvovec versus external control cohort). Covariates in the logistic regression model included the following baseline characteristics: age; NSAA total score; rise from floor velocity (/s); and 10-m run/walk velocity (m s−1).
In the estimation of the treatment effects, PS were used to adjust for differences between fordadistrogene movaparvovec-treated and external control participants using IPTW-ATT among treated participants. The IPTW-ATT weighting was calculated as follows:
$$\begin{array}{l}{Wi}=1\,{\text{if the}}\,{i}^{{th}}\,{\text{participant is in the high-dose}}\\ \qquad \quad{\text{fordadistrogene movaparvovec group}}\end{array}$$
$${Wi}={Pi}/(1-{Pi})\,{\mathrm{if}}\;{\mathrm{the}}\,{i}^{{\mathrm{th}}}\,{\mathrm{participant}}\;{\mathrm{is}}\;{\mathrm{in}}\;{\mathrm{the}}\;{\mathrm{external}}\;{\mathrm{control}}\;{\mathrm{cohort}}$$
where Pi is the probability of receiving fordadistrogene movaparvovec, conditional on the observed covariates. This creates a pseudo-control group that represents a random sample without any confounders. With these weights, fordadistrogene movaparvovec-treated participants received a weight of 1, and the external control participants received a weight of the odds of receiving fordadistrogene movaparvovec treatment. Thus, the population of fordadistrogene movaparvovec-treated participants serves as the reference population against which each of the fordadistrogene movaparvovec-treated and external control populations were standardized. If a participant in the external control cohort had a very high PS, a very large weight could be created. To reduce the impact of extreme weights, all weights with a value above the 95th percentile in the external control cohort were set equal to the 95th percentile. After trimming, the weights in the external control cohort were restandardized by dividing each individual weight by the sum of the total of weights in the external control cohort. After the PS weights were calculated, a linear model with a factor for treatment group was constructed to allow formal statistical comparisons between the fordadistrogene movaparvovec-treated and external control groups. In addition, doubly robust augmented inverse propensity weighting (AIPW) was performed6. The AIPW weights were included in the outcome regression model, using the treatment indicator and the same confounders (that is, the baseline covariates included in the PS model) to account for a doubly robust estimation.
The overlap of the distribution of the PS between fordadistrogene movaparvovec-treated and external control participants was visually inspected. For the PS weighting (IPTW-ATT) and doubly robust (AIPW) methods, the mean change from baseline for each treatment group along with the bootstrap s.e. and the bootstrap-bias-corrected 95% CI were provided. Additionally, the mean treatment group difference, associated bootstrap s.e., bootstrap-bias-corrected 95% CI and P value were provided.
A supplementary analysis plan for the external control analyses was created and finalized before the 1-year database lock.
Statistics and reproducibility
The sample size of this ongoing, nonrandomized, open-label, ascending-dose, phase 1b study was based on clinical (rather than statistical) considerations to provide adequate safety, tolerability and pharmacodynamic data. No statistical method was used to predetermine sample size. No data were excluded from the analyses. The experiments were not randomized. The investigators were not blinded to allocation during the experiments and outcome assessment. Given that this is a phase 1b, open-label, single-arm study with a small sample size, reproducibility may be constrained in the traditional sense because of the lack of a control group and the small sample size. However, standard clinical trial protocols and rigorous data collection were implemented, including prespecified endpoints to ensure the integrity and reliability of the results. In addition, an external control analysis was conducted, which allowed comparison of the findings with data from similar populations. All methods and data are fully documented, enabling future replication and validation of the findings. There was no formal hypothesis testing in this study according to the original design of this first-in-human study. Nominal P values are reported. The 95% CIs for changes from baseline in secondary (dystrophin expression and distribution) and exploratory functional endpoints were calculated and interpreted as descriptive statistics.
The primary analyses of safety outcomes included all enrolled participants who received fordadistrogene movaparvovec. Analyses of dystrophin expression included all participants who received fordadistrogene movaparvovec and had baseline and at least one post-dose dystrophin parameter of interest reported. Descriptive statistics were calculated for the incidence of AEs, laboratory values and changes in dystrophin expression levels according to the fordadistrogene movaparvovec dose level. All analyses used observed data without imputation for missing data unless specified otherwise. Assay results below the lower limit of quantification were imputed as 0.5× the lower limit of quantification, assuming uniform distribution, for summary statistics and graphical presentation. The missing steroid regimen starting date in the external control cohort was imputed as 6 months before the screening day per the eligibility criteria of the previous interventional trials.
To contextualize treatment effects observed with high-dose fordadistrogene movaparvovec on the exploratory functional endpoints, an external control cohort was derived from 156 placebo-treated participants with DMD from two previous interventional trials14,15 The external control cohort used for the analysis of the 1-year exploratory efficacy endpoints was compiled from two previous DMD studies15,32, which were chosen based on their recency, reflection of the current standard of care, randomized controlled design, data provenance and data quality. An internal independent team was created to validate the external control cohort in a non-biased manner to ensure the interpretability of the results and the validity of the conclusions. This team included a clinician and a statistician within Pfizer who were not part of the NCT03362502 (that is, the current) study team, were not directly involved with the day-to-day activities of the study team and did not have access to post-baseline outcome data.
To minimize selection bias, the NCT03362502 eligibility criteria, including age, ambulatory status, glucocorticoid use, the ability to rise from the floor within 7 s and cardiac function (data permitting), along with a requirement for nonmissing NSAA scores at baseline and at 12 months, were applied to create a subset of participants for the external control cohort.
PS adjustment methods were used to balance measured characteristics between ambulatory high-dose fordadistrogene movaparvovec participants and the external control cohort. The average causal treatment effect for high-dose-fordadistrogene movaparvovec-treated participants was estimated using IPTW-ATT. Prespecified covariates (that is, screening age, baseline NSAA total score, rise from floor velocity and 10-m run/walk velocity) were included in the PS model. Candidate covariates were selected based on their potential to influence treatment group selection or to be related to outcome risk. To address the potential over-influence of large weights, all weights above the 95th percentile in the external control cohort were truncated to the 95th percentile and then normalized according to the total of the weights in the group. Functional endpoints were analyzed using a linear regression model with treatment indicator and IPTW-ATT weights to adjust for differences between the high-dose fordadistrogene movaparvovec and external control groups. To evaluate the robustness of the findings, sensitivity analyses were conducted using doubly robust estimation to account for covariates in the outcome models. Bootstrap methods were used to obtain the estimated s.e. for the treatment effect and 95% CI without making underlying distributional assumptions. Balance of baseline variables between fordadistrogene movaparvovec-treated and external control participants in the unweighted and PS-weighted samples was examined using the standardized mean difference to quantify the magnitude of the difference between the two groups. The detailed methods used to derive and analyze the external control cohort can be found in the Methods.
Data as of 30 September 2022 were analyzed using SAS v.9.4 (SAS Institute) or R v. 4.1.3 (R Foundation)39.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.