TSC1 patients
The TSC1 group included 31 patients (15 males, 16 females), with a mean age of 25.1 years (range: 2–60 years). Of these, 45.1% had inherited variants, while 54.9% were sporadic or de novo. Pathogenic TSC1 variants were widely distributed throughout the gene, involving in particular exons 4, 5, 6, 10, 15, 17, 18, 21 and 22. One patient carried an intronic variant (intron 1). Variants were intragenic deletions (48.4%), nonsense (38.7%), missense (9.6%), and in one patient a duplication (3.2%) (Supplementary Table 1). Among these patients, six came from the same family and were affected by the already reported c.1498 C > T, p.(Arg500Ter) pathogenic variant (Fig. 1a).
Fig. 1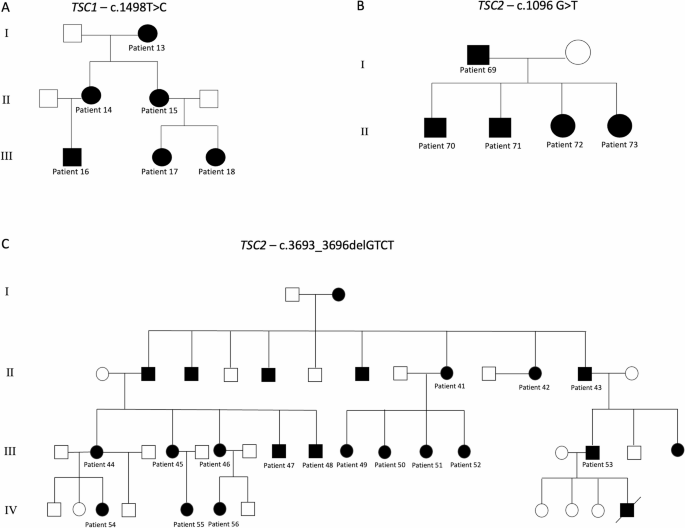
Genealogic tree of the three large families reported in the present study. (A) A family affected by mutation c.1498T > C of TSC1; (B) A family showing c.1096 G > T mutation of TSC2; (C) A large family affected by mutation c.3693_3696delGTCT of TSC2. Patients affected by TSC are marked in black.
TSC1 patients were affected by different types of seizures, differing in EEG findings, clinical course, age of onset and type of seizures. Epileptic seizures were present in 64.5% of patients, with a mean onset at 35.4 months. Seizure types were both generalized and focal seizures and some patients experienced multiple seizure types throughout life. More in detail, two patients (6.4%) presented infantile spasms, 12 (38.7%) had focal seizures (three with impaired awareness), six (19.3%) patients experienced generalized tonic clonic seizures, four (12.9%) suffered absence, one (3.2%) presented gelastic seizures, one (3.2%) reported seizures with fever, three (9.6%) suffered hypertonic seizures and one (3.2%) experienced atonic seizures. Their frequency ranged from 1 per lifetime to 28 per week (mean: 10.4 weeks, SD: 9.2). Severity and duration of seizures varied among patients and in the same patient. EEG abnormalities included spikes, waves, and hypsarrhythmia. Particularly, 11 patients (35.5%) had normal EEG, one (3.2%) presented hypsarrhythmia, 13 (41.9%) were reported to have spikes and waves (unilateral or bilateral), 11 (35.5%) had synchronous or asynchronous focal spikes, one (3.2%) had “arceau-like” pattern, four (12.9%) presented a theta wave pattern. Epilepsy course varied from spontaneous remission to drug resistance, with valproic acid and carbamazepine being the most common treatments: ten out of 20 patients (50%) achieved remission (complete response), while one (5%) had a good response, two (10%) a partial response, and the other and seven other (35%) showed a drug-resistant epilepsy.
Cognitive impairment levels of TSC1 patients ranged from mild to severe: 51.6% had normal cognitive functions, 9.7% had cognitive function near lower limit (borderline), 19.3% presented mild intellectual disability, 3.2% suffered from moderate intellectual disability and 16.1% had severe cognitive deficit. Autism spectrum disorder (ASD) was observed in four patients (12.9%), with only one patient showing Level 2 autism. Behavioral disorders (e.g., ADHD, anxious-depressive syndrome, borderline personality disorder) affected eight patients (25.8%), while 12 (38.7%) experienced learning disabilities. Sleep disorders were reported in only three patients (9.7%). Brain abnormalities of TSC1 group included cortical tubers (67.8% with > 5 tubers, mean size 8 mm) and subependymal nodules (51.6% with > 5 SENs, variable in size). Subependymal giant cell astrocytomas (SEGAs) were rare, found in only two patients (6.5%). White matter abnormalities and cerebral cysts were uncommon in our group of TSC1 patients, only one patient (3.2%) carrying the nonsense mutation c.2293 C > T, had many cerebral cysts.
Some pathogenic variants were associated with specific clinical patterns. The mutation c.1888_1891delAAAG, identified in two patients, was associated in both patients with numerous bilateral tubers and SENs, while epilepsy and neuropsychiatric disorders were heterogeneous, suggesting a potential role of other modifying factors. The pathogenic variants c.1004del and c.2111_2112del were linked to minimal neurological symptoms and a good quality of life, indicating a milder impact. The c.1498T mutation (associated to a common specific EEG pattern and many brain hamartomas) was found in 6 patients of the same family (Fig. 1a) and five of them showed the same EEG pattern (sharp theta waves and spikes in the left temporal lobe or bilateral). Notably, in this family, not all the patient showed epilepsy, and some of those affected were partial responders to drugs, while other drug-resistant. Cognitive functions and behavioral disorders were variable, with different grade of intellectual disability (absent to moderate/severe).
TSC2 patients
This study analyzed 50 patients with pathogenic TSC2 variants, comprising 21 males (42%) and 29 females (58%), with a mean age of 27.1 years (SD = 18.6, range 2–71 years). Variants were classified as familial (23 cases, 46%) or sporadic or de novo (27 cases, 54%) and showed extensive distribution across exons and included deletion (60%), nonsense (26%), missense (10%), and splicing mutations (6%). Furthermore, four intragenic deletions were detected in this group (one spanning from intron 1 to exon 8), as well as somatic mosaicism for the TSC2 c.3598 C > T (p.Arg1200Trp) variant, identified with a variant allele fraction of 11.4%. Among these patients, two large families were included: one (five members) harbored the c.1096G > T; (p.Glu366*) mutation (Fig. 1b). The other one comprised 16 members (among the 50 known members of this family), and presented the already reported c.3693_3696del (p.Ser1232Thrfs*92) mutation (Fig. 1c).
Of the 50 patients, 36 (72%) experienced seizures, with a mean onset age of 29.45 months (SD = 66.3, range one month to 23 years). Seizure types varied, with 32% experiencing infantile spasms, 30% focal seizures (16% with impaired awareness) and 22% generalized tonic-clonic seizures. The frequency and severity of seizures varied widely, ranging from two lifetime episodes to as many as 42 episodes per week (mean: 18.3; SD: 24.5). EEG patterns were varied, including theta waves, spikes, hypsarrhythmia, and focal spikes. Eighteen patients (36%) had normal EEG results, while focal and generalized spikes were more frequently observed. EEG patterns fluctuated over time within individual patients. The clinical course of epilepsy in TSC2 group of patients was variable, going from remission to multidrug resistance. Different antiseizures medications were used, including valproic acid, vigabatrin, carbamazepine, ACTH, topiramate, phenobarbital, levetiracetam, oxcarbazepine, lamotrigine, everolimus. Among them the most used were valproic acid and carbamazepine. TSC2 patients had been divided according to drug response into complete responder, (13 out of 36 patients, 36.1%), good responders (four patients, 11%), partial responders (six patients, 16.6%) and non-responders (nine patients, 25%).
Cognitive impairment was prevalent among TSC2 patients, with 24%, 10%, and 24% exhibiting mild, moderate, and severe intellectual disability, respectively, while 42% displaying normal cognitive function. ASD was diagnosed in 12% of patients, while 48% had learning disabilities. Behavioral issues were present in 30% of patients, predominantly ADHD (20%), irritability (10%), and oppositional behavior (10%). It should be noted that many patients presented more than one behavior disorder. Sleep disorders were rare, with only a few cases of insomnia or enuresis reported. MRI analyses revealed that 10% of TSC2 patients had no detectable cortical tubers, while 74% had multiple tubers (> 5). The average tuber sizes was 17 mm (SD = 18.3 mm), with some patients presenting cerebellar or cystic tubers. Subependymal nodules (SENs) were identified in 86% of patients, with dimensions varying widely (average size = 9.4 mm; SD = 0.9 mm). SEGAs were less common, found in 14% of patients. The average size of SEGAs was 13 mm (SD = 15 mm), ranging from 1 cm to 1.5 cm. In our cohort SEGAs were associated with different genotypes, in particular the pathogenic variants c.3094 C > T, c.5201_5216dup and c.1283_1285del. One patient harboring 2 SEGAs carried a large intragenic deletion (spanning from exon 3 to 9). Radial bands were noted in 12% of cases, mostly in parietal and temporal regions. Most patients (74%) had no white matter abnormalities, the remaining cases ranged from multiple to unspecified abnormalities. Only 3 patients (6%) had brain cysts and 2 (4%) had atrophy or hypoplasia of the corpus callosum.
Regarding the specific pathogenic variant c.3693_3696del, (p.Ser1232Thrfs*92), it was found in one of the biggest family of TSC patients in Italy. The family is made up of at least 43 members (Fig. 1c), of whom at least 25 members are affected by tuberous sclerosis. Complete information about genotype and phenotype of all the members was impossible to collect for many reasons (for example, some of them died without certain diagnosis, others refused clinical and genetic investigations, data were stored in different hospitals): for this reason, a total of 16 family members have been included in this study. Although they all carry the same mutation, an intrafamilial variable clinical presentation was observed, going from mild cutaneous phenotype to severe mental retardation and drug-resistant epilepsy. Nine patients from this family presented epilepsy. The median age of onset of epilepsy was 6 years old, ranging from birth to 23 years. Types of seizures and frequency were variable, but the most frequent were infantile spasms, tonic clonic and focal crises with impaired awareness. Regarding the EEG, the most frequent patterns were hypsarrhythmia and focal spikes (with secondary generalization) in the left frontal temporal lobes: it must be underlined that all the normal EEGs found in TSC2 epileptic patients were observed in this family. Drug response was different among the members of this family, and the biggest group is made up of good/partial responders, although there were four cases of remission. All the levels of cognitive impairment severity were present in this family, although the most frequent was mild intellectual disability. ASD was rare and none of the individuals met the full diagnostic criteria for confirmed diagnosis, only some of them had a subclinical features of ASD with limited social difficulties. Learning disability was present mainly in patients with moderate-severe intellectual disability, who were the minority of the family. The most frequent behavior disorders were ADHD and oppositional disorder. Sleep disorders were basically absent. Many bilateral tubers and SENs were reported in all the members of the family, while none of them presented SEGAs. Cerebral cysts and white matter abnormalities were absent, while some of them presented radial bands. To sum up the common characteristics of this family members were multiple tubers and SENs, absence of SEGAs, attention deficit disorder, mild developmental delay (that could explain why they managed to reproduce), partial epilepsy control, infantile spasms and focal impaired awareness seizures.
The pathogenic variant c.1096G > T (p.Glu366Ter) was present in another family (patients 69,70,71,72,73) (respectively a father and his two sons and two daughters). The types of seizures described in this family were focal with impaired awareness and infantile spasms. All patients also shared severe intellectual and learning disability. ASD, behavior and sleep disorders were absent. Brain MRI reports revealed the presence of few to many tubers and SEN, but no-one showed SEGA.
TSC1 vs. TSC2 patients
The study cohort included 81 patients, divided into TSC1 and TSC2 groups. Tables 1, 2 and 3 summarize comparisons between the two groups and the statistical data.
Table 1 Main clinical characteristics of the two groups of patients.Table 2 Epilepsy and neuropsychiatric comorbidities of the two groups of patients (included if at least 5 individuals presented the specific feature) * Patient 9, 44 and 49 – experiencing only one to two seizures in their lifetime – are excluded from this range.Table 3 MRI finding of the two groups of patients. SD: standard deviation; SEGA: subependymal giant cell astrocytoma.
The proportion of TSC2 patients (61.7%) was higher than TSC1 ones (38.3%). There was no statistically significant difference between the two groups with respect to sex (TSC1: male 48.4%-female 51.6%; TSC2: male 42%-female 58%) or mean age. In our cohort, the distribution of familial versus sporadic or de novo pathogenic variants was comparable between the TSC1 and TSC2 groups. There was no statistically significant difference for types of mutations, which included intragenic deletions, duplications, missense and nonsense mutations.
TSC2 patients exhibited a greater variability in clinical manifestations, seizure types, cognitive outcomes, and behavioral disorders. There was no statistically significant difference in the overall prevalence of epilepsy between the two groups (72% in the TSC2 group vs. 64.5% in TSC1), but infantile spasms were significantly more frequently in TSC2 patients (p = 0.016). For other seizure types (focal, focal dyscognitive, tonic-clonic, atonic, hypertonic, myoclonic, gelastic) no statistically significant differences were observed. EEG findings were comparable between TSC1 and TSC2 patients, with a similar proportion showing normal patterns. Pathological findings – such as focal or generalized spike-and-wave discharges and hypsarrhythmia – were also observed at similar rates in both groups, with no significant differences in their prevalence.
Both groups showed similar responses to antiseizure medications, with no significant difference in rates of remission, partial or good response, or drug-resistance. Cognitive and behavioral disorders were similarly distributed. Importantly, seizure remission was associated with a higher likelihood of normal cognitive function. Among the TSC1 patients with seizure remission, 70% had normal cognition (90% if those with mild impairment were included). Among TSC2 patients with remission, 28.6% had normal cognitive profile, rising to 78.6% when including those with mild intellectual disability.
Tubers, SENs, and radial bands occurred at comparable frequencies, although TSC2 patients slightly more likely to have multiple and larger tubers (p p TSC1 patients were more likely to present radial (p p