Selection and integration of candidate genes governing silique and seed size
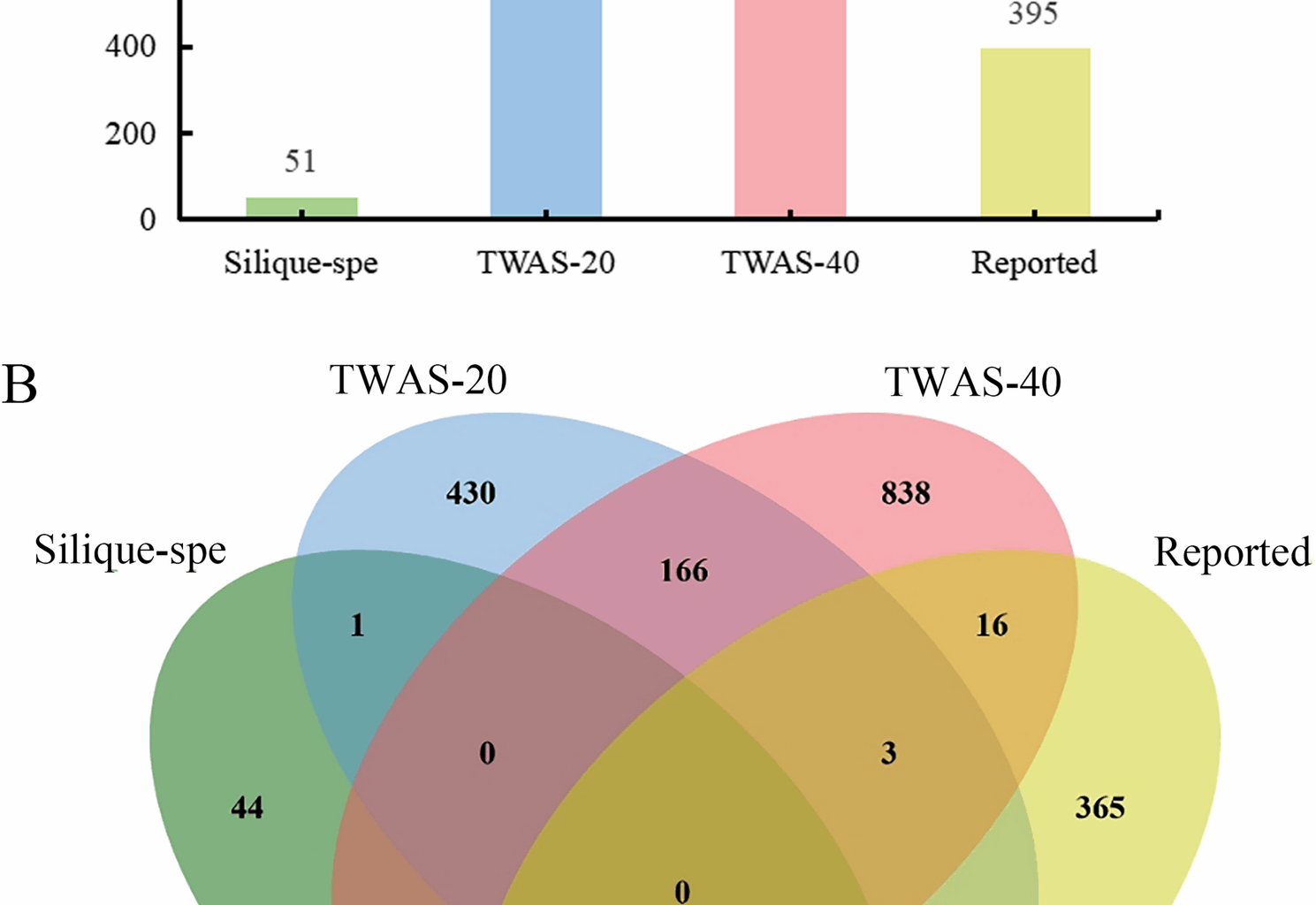
To identify candidate genes modulating silique length and seed size in B. napus, we first interrogated TWAS data from various developmental stages of siliques [38]. We focused on genes exhibiting highly differential expression patterns and strong correlations with traits such as oil content, protein content, hull percentage, and TSW, ultimately identifying 1472 candidate genes potentially regulating silique length and seed size (Fig. 1A, B). Complementing this approach, we synthesized a compendium of 395 functionally related genes from literature reports on model plants like Arabidopsis thaliana and rice, focusing on seed size, floral organ size and TSW. Additionally, through transcriptome analysis of Zhongshuang 11 (ZS11), we isolated 89 silique/silique wall-specific expressed genes, corresponding to 51 orthologs in Arabidopsis thaliana (Fig. 1A, B).
Upon eliminating redundant genes from these various sources, we compiled a comprehensive list of 1905 candidate genes related to silique and seed size. Notably, the genes identified through TWAS displayed minimal overlap with previously reported genes, indicating that the mechanisms governing silique development and seed size regulation in B. napus may diverge from those in model plants. This mutant library holds significant value for studying silique development and seed size traits in B. napus (Fig. 1).
Fig. 1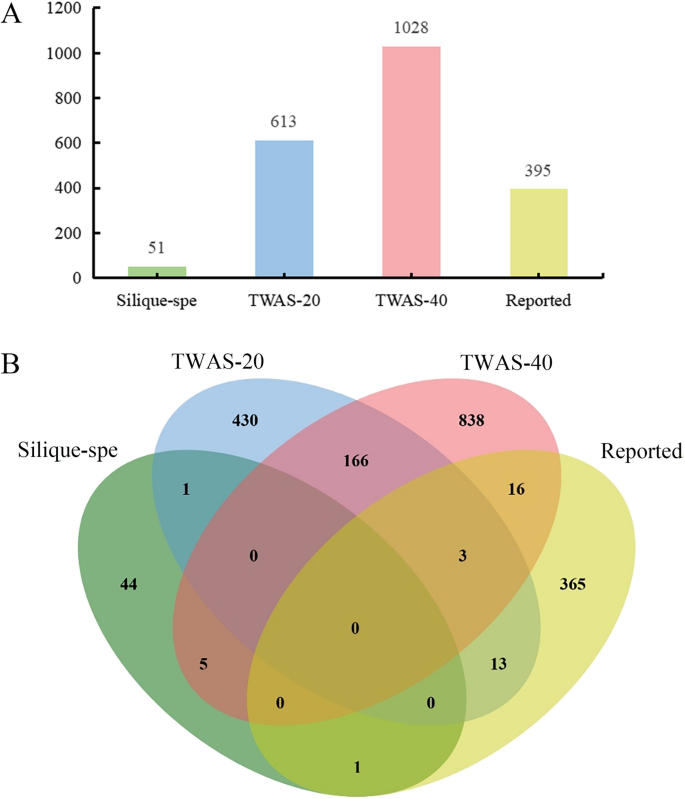
Candidate gene screening from different sources. A Candidate gene from diverse sources; B Integrative comparison of candidate genes derived from different sources. TWAS-20 and TWAS-40 represent differently expressed candidate genes associated with the traits such as oil content at 20 days after flowering and 40 days after flowering. Silique-spe represents candidate genes specifically expressed in silique and seed. Reported represents genes associated with silique or seed size have been reported in plants
Creation and quality assessment of a mutant library for candidate genes related to silique and seed size
We performed BLAST analysis on the 1,905 candidate genes using the XiaoYun information website (http://yanglab.hzau.edu.cn/BnGDXY/#/) to obtain all the homologous copies and gene sequence information of the candidate genes. Our analysis revealed that 1,691 of these genes possessed four or fewer homologous copies, whereas 214 genes exhibited five or more copies (Table 1). Given the technical challenges associated with generating homozygous mutations across all copies of multi-copy genes, we prioritized the design of sgRNAs targeting candidate genes with four or fewer homologs. This strategy ensured a more focused and feasible approach to creating a robust mutant library for functional analysis.
Table 1 Homologous information collation of candidate genes
Drawing upon the homologue gene copies and sequence data of the candidate genes, we employed the CRISPR-P 2.0 sgRNA online design tool (http://crispr.hzau.edu.cn/CRISPR2/) to curate an sgRNA library. In total, we designed 6,124 sgRNAs, targeting 1,739 candidate genes, with an average of 3.52 sgRNAs per gene, thereby ensuring the potential for editing all copies of the targeted genes. Notably, certain sgRNAs exhibited the capacity to target up to six homologous copies. A subset of the sgRNA details is presented in Table S2.
Utilizing the Oligo array method, we conducted large-scale synthesis of the sgRNA library. The synthesized sgRNAs were ligated to the vector plasmid via a mixed ligation approach, followed by transformation into E. coli (DH5α) and plasmid extraction, ultimately yielding a mixed plasmid library (Fig. S1). To assess the quality of this library, we examined the accuracy, coverage, and uniformity of sgRNA sequences within the plasmid library. Based on the vector sequence information, we designed universal PCR primers (Table S1) proximal to the sgRNA insertion site and conducted PCR using the purified plasmids as templates. Following purification, the PCR products were sequenced using an Illumina X-Ten sequencer to obtain sequence information for subsequent analyses. The results showed that 6,122 of the 6,124 designed sgRNAs were successfully synthesized, achieving a coverage of 99.90% and a deviation value (the ratio of the highest reads count to the lowest reads count) of 4.94 (Fig. 2). This indicates that the excellent coverage and uniformity of the plasmid library. Subsequently, the plasmid library was transformed into Agrobacterium tumefaciens (GV3101) using electroporation, and the quality of the resulting Agrobacterium library was also assessed. The analysis indicated that 6,120 sgRNAs were detected in the Agrobacterium library, maintaining a coverage of 99.93% and a deviation value of 4.53 for sgRNA distribution (Fig. 2). These findings confirm that the plasmid library retained its high coverage and uniformity following transformation into Agrobacterium.
Fig. 2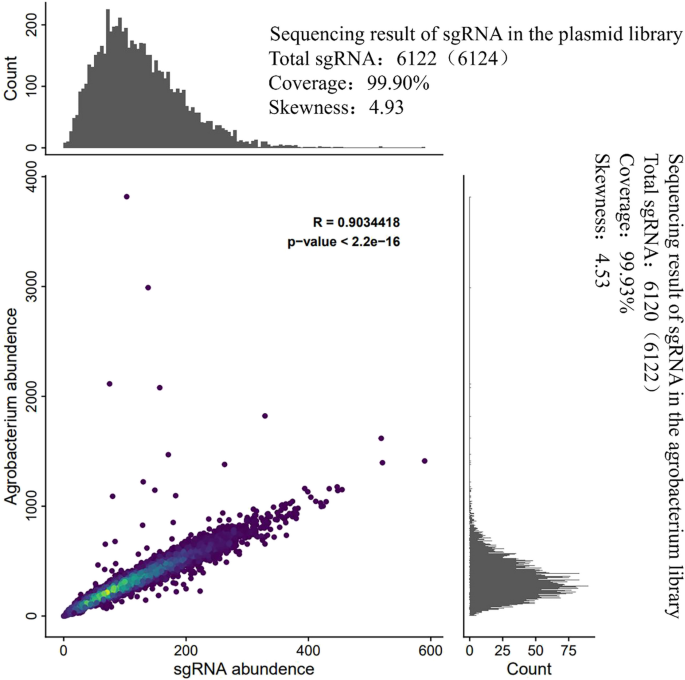
Comparison of Quality Testing Results of the sgRNA plasmid library
Random transformation of mutant library
Utilizing our optimized Agrobacterium-mediated hypocotyl transformation system, we successfully transferred the plasmid library into the rapeseed, yielding a total of 681 regenerated plants. Among 621 T0 plants screened, 567 (91.3%) were confirmed as transgenic positives (Fig. S2). Subsequently, we employed high-throughput techniques to profile the sgRNAs in regenerated individuals (Table S3). Among the 681 T0 generation individuals, 453 sgRNAs were detected. 71.51% of the T0 generation individuals harbored a single sgRNA, 20.85% carried two sgRNAs, while only 3.38% and 1.17% had three and four sgRNAs, respectively (Fig. 3A). Of the 487 single-sgRNA transformed T0 individuals, a diverse of 281 sgRNAs was identified, of which 92.53% of the sgRNAs were found in 1 to 3 independent T0 individuals (Fig. 3B). Strikingly, 92.53% of these sgRNAs were detected in 1 to 3 independent T0 individuals (Fig. 3B). This observation suggests a high degree of uniformity in the mixed library during the Agrobacterium-mediated hypocotyl transformation of B. napus.
Fig. 3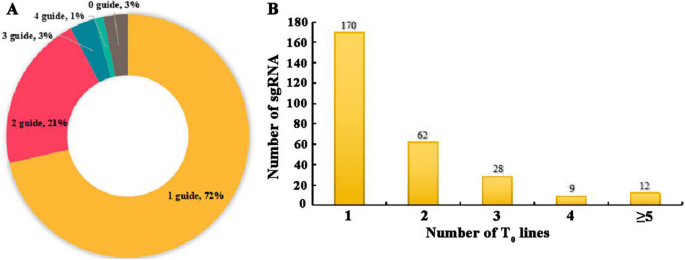
sgRNA typing of T0 transformed plants. A The status of sgRNA in T0 transformed plants. B Analysis of T0 plants containing a single sgRNA
Analysis of the genetic editing status in T0 plants
Based on sgRNA genotyping results, candidate gene-specific editing detection primers were designed, and the gene targeting editing status was analyzed through high-throughput techniques (Table S4 and S5). Out of 408 tested individuals, 151 plants (37.00%) exhibited targeted gene editing (Fig. 4A). Among these, nine T0 plants underwent two targeted gene editing events, two T0 plants underwent three targeted gene editing events, one T0 plant underwent four targeted gene editing events, and the remaining 139 T0 plants showed one targeted gene editing event (Fig. 4B). Subsequently, we assessed the efficiency of different sgRNAs. Among the 249 sgRNAs analyzed, 128 sgRNAs (51.41%) exhibited editing activity (Fig. 4C). Currently, the 151 genetically edited plants target 84 candidate genes. These genes include 71 selected from TWAS data during various developmental stages of the silique, 11 derived from existing literature, and 2 specifically expressed in the silique of B. napus (Fig. 4D).
Fig. 4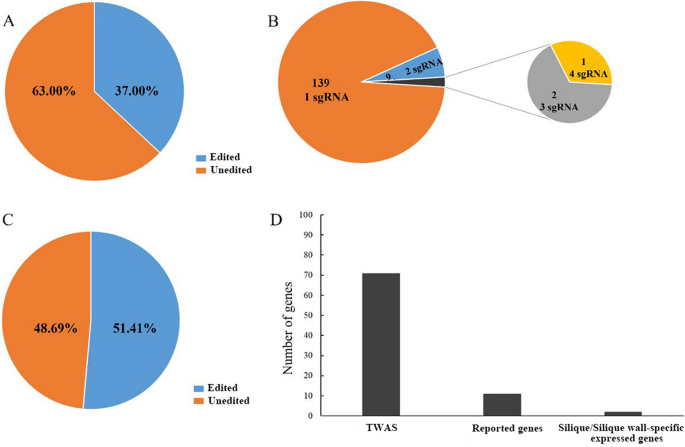
Analysis of editing efficiency. A The edited rate of T0 transformed plants. B Analysis of sgRNA in T0 generation edited plants. C The edited rate of sgRNA in T0 transformed plants. D The candidate gene sources of T0 generation edited plants
Based on the editing detection results of the T0 generation plants, the CRISPR/Cas9 system used in this study produced multiple editing types in candidate genes, including single-base insertions or deletions, base substitutions, large fragment insertions or deletions, and mutations with several different editing types occurring at the same site (Table 2). Further analysis of all editing types revealed that the editing types were dominated by single-base insertions, followed by multi-base deletions (Fig. 5A). Additionally, there was a significant preference for single-base insertion edits (Fig. 5B), and the frequency of insertion of different bases from highest to lowest was: adenine (A) > thymine (T) > cytosine (C) > guanine (G).
Fig. 5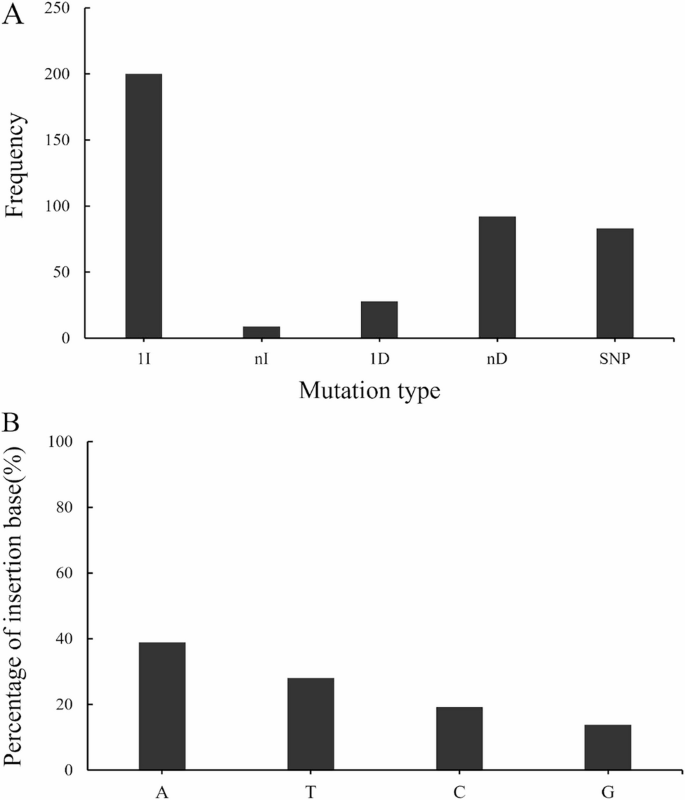
Analysis of Edit the type of T0 generation edited plants. A The edited type of T0 generation edited plants. SNP: Base substitution; 1I: one base insertion; nI: multiple base insertion; 1D: one base deletion; nD: multiple base deletion. B Frequency of different bases for 1 bp insertion
Table 2 The editing type in partial T0 generation plantsEditing type analysis in T1 generation edited plants
Most of the T0 generation edited plants are heterozygous or chimeric edited individuals, necessitating further propagation to isolate homozygous mutant lines (Table S5). We propagated the self-fertilized progeny of 19 selected T0 lines on a comprehensive breeding platform, including A03, A21, A29, A49, A60, A68, A73, A75, A79, B27, B29, B48, B49, B78, B83, B85, C37, D27, and D35. Subsequently, we employed Hi-Tom sequencing platform to analyze the editing status of the offspring of these lines.
Our findings revealed a robust transmission of editing events from the T0 generation to the subsequent T1 generation, with heterozygous and chimeric genotypes undergoing segregation processes that ultimately led to the emergence of homozygous mutants (Fig. 6A). Notably, within the T1 lines of A21, B29, and B49, we successfully isolated homozygous individuals, each harboring double alleles that had undergone precise editing (Table S5). Importantly, the sgRNAs utilized in these three T1 lines effectively targeted both homologous copies of the respective genes, confirming their status as fully edited plants.
Fig. 6
The editing status in partial T1 generation mutants. Dashes (−) represents a missing base; Red font represents base insertion; Blue font represents base substitution; WT represents a wild-type sequence
In the T1 generation lines designated A68, A73, and A79, their corresponding sgRNAs only targeted one homologous copy of the corresponding gene (Table S5). For instance, in the T1-A73-08 plant, sgRNA-0432 exclusively targeted BnaA09G0426100XY (homologous to AT1G24150) achieving homozygosity. However, given the presence of an additional homologous copy in Xiaoyun. This individual necessitates subsequent crossing with a homozygous mutant of BnaC05G0208300XY to secure a double-allele homozygous mutant. Similarly, line A03 yielded a homozygous double-allele mutant targeting BnaA03G0180700XY and BnaC03G0200300XY (homologs of AT2G36450), yet editing was absent in BnaA04G0238800XY and BnaC04G0552700XY. Consequently, hybridization screening is imperative to isolate homozygous mutants encompassing all four gene copies.
Conversely, in the T1 lines of A49, A29, and B27, despite sgRNAs intended to target all homologous gene copies, distinct copies underwent homozygous editing in separate individuals (Table S5). Illustratively, sgRNA-1573 in the T0-A49 individual aimed at two homologs of AT2G43120 in Xiaoyun: BnaC03G0244300XY and BnaA03G0216100XY. In T1-A49-13, homozygous editing was confined to BnaA03G0216100XY, whereas T1-A49-24 exhibited homozygosity in BnaC03G0244300XY. To isolate homozygous double-allele mutants for both copies, continuous self-crossing and purification of T1-A49-13 and T1-A49-24 lines are being pursued.
Comprehensive analysis of the detection results revealed that the occurrence of base variations consistently localizes 3 bp upstream of the PAM sequence, corroborating prior investigations. Furthermore, within select T1 generation individuals, we uncovered novel editing modalities. For instance, the T1-B83-1 line exhibited a new editing pattern in the BnaA01G0293900XY copy, featuring a transformation from a 3 bp deletion to a 4 bp deletion (Fig. 6B). Similarly, in the T1-A79-2 line, we observed a distinct single-base insertion event, with an adenine inserted at the third base before the PAM sequence (Fig. 6C). These emergent editing patterns may stem from the protracted activity of Cas9 protein at the targeted genomic loci.
In summary, through successive generations of purification, homozygous edited lines have been obtained in 10 T1 generation lines, including A03, A21, B29, B49, A68, A73, A79, A49, A29, and B27. The edited genes are homologous to genes such as AT2G36450, AT5G57790, AT5G47330, AT3G52115, AT5G52050, AT1G24150, AT5G52600, AT2G36450, and AT2G43120.
Bioinformatics analysis of the BnHRDs genes
AT2G36450 encodes an Integrase-type DNA-binding superfamily protein HRD, a member of the DREB subfamily A-4 within the ERF/AP2 transcription factor family. This nuclear-localized protein harbors a conserved AP2 domain and exhibits a broad expression profile across A. thaliana tissues, including stems, leaves, inflorescences, petals, developing siliques, and seeds, with heightened levels in stems, pollen, and developing seed (data from TAIR). Notably, ectopic overexpression of this gene potentiates root network elaboration and fortifies resilience against water and salt stress [43].
Utilizing the BnIR database, a comparative analysis of the expression patterns of homologous genes for BnaHRDs in ZS11 tissues at various developmental stages revealed distinct expression signatures, primarily in roots, stems, and developing siliques. These patterns deviate from those observed in A. thaliana, and further intra-species variations were discernible among different gene copies (Fig. S3). Four homologous copies of this gene were detected in the Xiaoyun genome: BnaHRD.A03 (BnaA03G0180700XY), BnaHRD.A04 (BnaA04G0238800XY), BnaHRD.C03 (BnaC03G0200300XY), and BnaHRD.C04 (BnaC04G0552700XY). Predicted protein products of AtHRD and its BnaHRD counterparts vary slightly in length, comprising 184, 187, 179, 187, and 176 amino acids, respectively, yet they exhibit remarkable conservation within the AP2 domain (Fig. S4A). Sequence homology analysis using Geneious software revealed high sequence similarities between AtHRD and its BnaHRD counterparts, Ranging from 77.9 to 80.5%, underscoring their evolutionary relatedness. To gain further insights into their functional motifs, we employed MEME and TomTom tools to conduct a comprehensive motif scanning and annotation. This analysis identified six distinct motifs in AtHRD and its BnaHRD homologs, with four motifs being universally present across all five proteins, indicating shared functional attributes (Fig. S4B). ProParam predictions also indicate similar physicochemical properties between AtHRD and BnaHRDs (Table S6). These results imply that the core functional attributes of BnaHRDs closely resemble those of AtHRD, but subtle functional divergences may potentially arise among distinct BnaHRD copies, suggesting a degree of functional diversification within this protein family.
Analysis of mutation types and phenotypes in BnaHRDs homozygous mutants
Currently, our efforts have yielded targeted mutant lines exclusively for BnaHRD.A03 and BnaHRD.C03, from which we have meticulously screened and characterized an array of homozygous mutant variants exhibiting diverse mutation profiles. In T1-A03-15 line, a 14-base deletion occurred in BnaHRD.A03, whereas a single-nucleotide insertion is evident in BnaHRD.C03. Conversely, the T1-A03-21 mutant displays a homozygous editing pattern solely in BnaHRD.A03, with BnaHRD.C03 manifesting a heterozygous genotype comprising a single-nucleotide insertion mutation alongside the wild-type sequence. Besides, the T1-A29-26 mutant harbors a 5-base deletion in BnaHRD.A03, while a 12-base deletion and a 1-base substitution in BnaHRD.C03 (Fig. 7A). Upon analyzing the amino acid sequences, we observed that the mutations in T1-A03-15, T1-A03-21, and T1-A29-26 led to premature translation termination in the proteins encoded by BnaHRD.A03, commencing at amino acid positions 118 and 122. Similarly, the mutations in T1-A03-15 and T1-A03-21 caused premature termination in the proteins encoded by BnaHRD.C03, starting at amino acid position 123. However, in T1-A29-26, the protein product encoded by BnaHRD.C03 does not experience premature termination but instead loses 4 amino acids at positions 113–116 (Fig. 7B).
Fig. 7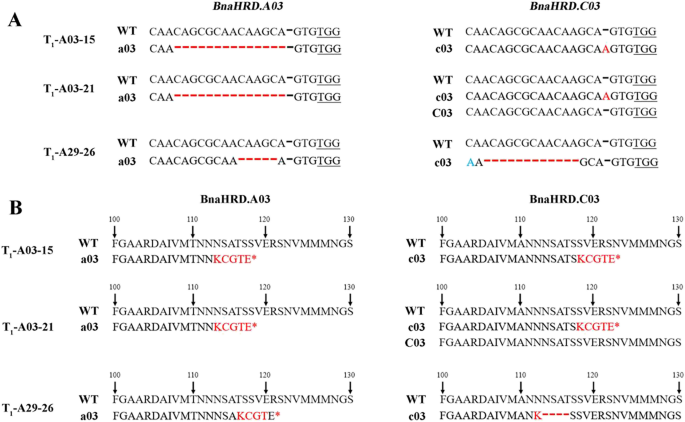
Editing and amino acid sequence analysis of BnaHRDs homozygous mutants. A Target editing of some BnaHRDs T1 homozygous mutants; B Amino acid sequence analysis of some BnaHRDs T1 homozygous mutants. WT represents the wild-type sequence, a03 represents the BnaHRD.A03 copy on chromosome A03, c03 represents the BnaHRD.C03 copy on chromosome C03, and C03 also represents the wild-type BnaHRD.C03 sequence
In comprehensive breeding platform, we cultivated these homozygous mutants and rigorously assessed their associated agronomic traits. The results revealed consistent yet nuanced trends in these traits, marked by reductions in PH, MIL, and SL, along with a decrease in ENS and an increase in SNPS, while TSW declined (Fig. 8). Notably, pronounced reductions in PH, MIL, and SL, coupled with a diminished ENS, were exclusive to the T1-A29-26 mutant line. Furthermore, significant augmentations in SNPS and TSW reductions were uniquely observed in the T1-A03-21 and T1-A03-15 lines, respectively (Fig. 8). The phenotypic variations observed among mutants may arise from allele-specific editing effects, reflecting functional divergence between homologous copies.
Fig. 8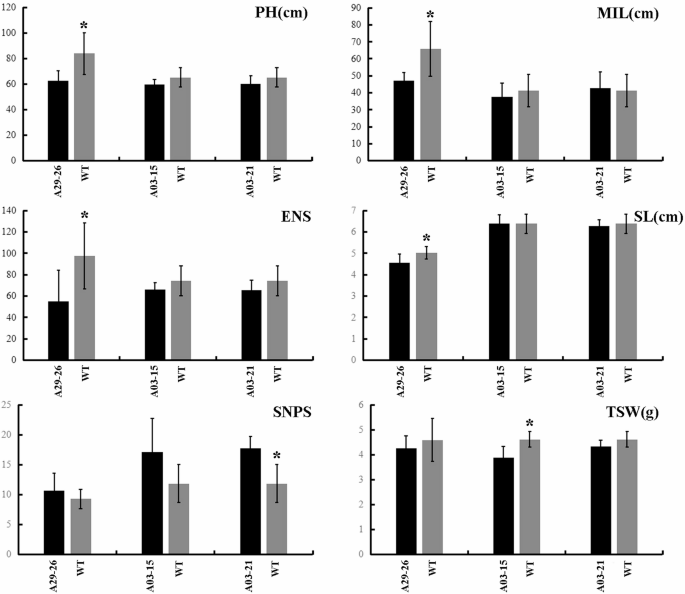
Analysis of Agronomic Traits of BnaHRDs Homozygous Mutant. PH Plant height, MIL Length of main inflorescence, ENS Effective number of siliques, SL Silique length, SNPS Number of seeds per silique, TSW Thousand seed weight. * p