Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) was first identified in late 2019. The subsequent pandemic has had substantial impacts on global patterns of virus activity. The abnormal prevalence of other respiratory viruses has prompted speculation regarding significant evolutionary shifts in viral genetic profiles, as viral genetic evolution has undergone substantial changes under the pressure of nonpharmaceutical interventions (NPIs) [20]. Globally, a 2022–2023 study conducted in Arizona, USA, documented mutations within the prefusion F protein antigenic sites of both the RSV-A and RSV-B subtypes. Sequencing analyses revealed multiple nonsynonymous substitutions in the antigenic domains of both subtypes. Given that RSV vaccine design is predicated on the prefusion conformation of the F protein, these mutational events may compromise the efficacy of RSV vaccines [21]. In China, research has indicated that the mutation rate of the influenza A (H1N1) virus notably increased during the COVID-19 pandemic. Concurrently, genetic reassortment events were documented among distinct lineages of the H3N2 virus, which further accelerated viral mutagenesis [22, 23]. On the basis of the above research status, we conducted complete-genome sequencing and genetic evolutionary analysis of HBoV that was circulating in Beijing in 2023 (the first year of COVID-19 prevention and control was managed as a Class B infectious disease in China) to elucidate the factors underlying the aberrant prevalence of respiratory viruses during the post-COVID-19 pandemic period.
HBoV is an emerging virus worldwide that is closely related to respiratory tract infections in children. As a nascent virus, the limited understanding of the epidemiological patterns and structural characteristics of HBoV has hindered its effective prevention, resulting in recurrent annual outbreaks of respiratory and gastrointestinal infections associated with this pathogen [7, 24, 25].
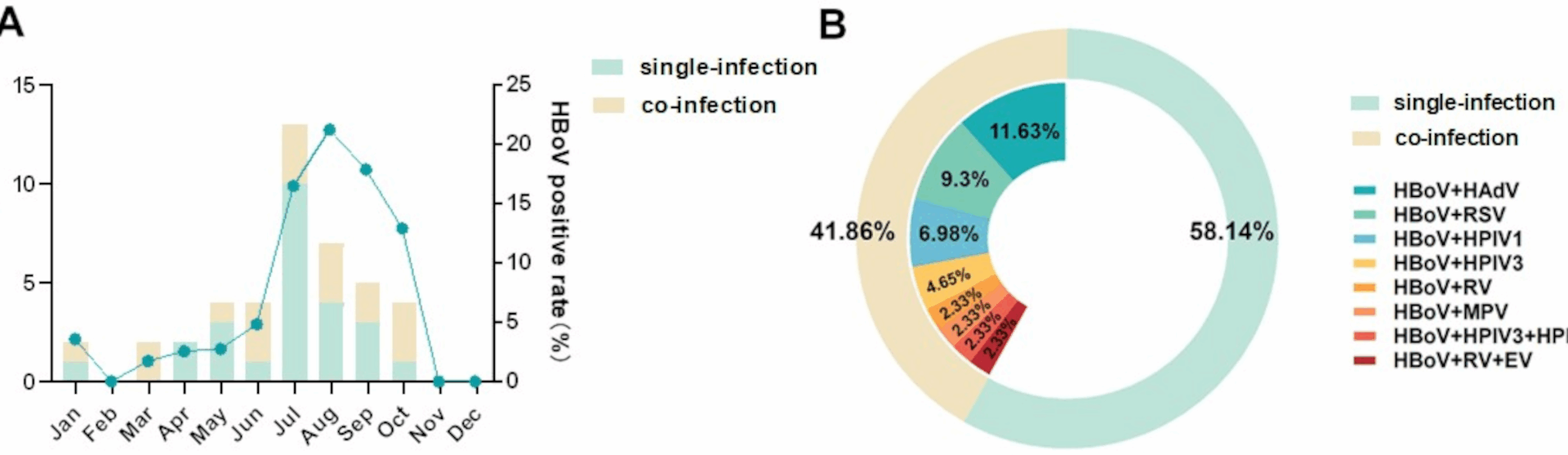
In this study, we analyzed the genetic characteristics of HBoV among outpatient and hospitalized children from three hospitals in Beijing from January to December 2023. The results demonstrated an HBoV-positive detection rate of 2.98%. No significant differences in demographic characteristics were observed in terms of age or sex among the infected population. Our study revealed a higher prevalence of HBoV during the summer months, whereas the existing literature shows inconsistent findings regarding its seasonal prevalence patterns in China [7, 25,26,27,28]. A Suzhou-based study demonstrated that the detection rate of HBoV exhibited no statistically significant variation before and after the COVID-19 pandemic, with postpandemic epidemiological trends of HBoV remaining consistent with historical patterns, which were more prevalent in summer and winter [29, 30]. As the HBoV case data from 2023 alone are insufficient to establish its seasonal epidemic patterns, the current study provides preliminary observational evidence rather than definitive conclusions. The seasonal frequency of HBoV infections should be systematically investigated in further studies.
High rates of pathogen coinfections represent a well-documented characteristic of HBoV infections. Epidemiological surveillance studies conducted across multiple Chinese regions, including Beijing, Kunming, and Lanzhou, have consistently reported elevated coinfection frequencies, with HBoV and RSV coinfections demonstrating a particular prevalence [7, 27, 28, 31,32,33,34]. In accordance with these findings, our data identified HAdV and RSV as the predominant pathogens coinfected with HBoV.
Phylogenetic analyses based on both the complete HBoV genome and the VP1 gene sequences consistently revealed four well-defined genetic subtypes. The remarkable congruence observed between the phylogenetic analyses derived from VP1 gene sequences and those obtained from whole-genome comparisons indicates that VP1 sequence analyses provide reliable molecular markers for inferring the evolutionary relationships among HBoV isolates. HBoV was initially identified in China in 2006, becoming the third country, after Sweden and Australia, to report confirmed HBoV infections [3]. The virus has since become endemic in China. Phylogenetic analysis demonstrated that the majority of HBoV1 isolates identified in mainland China formed a distinct cluster. Notably, isolate PV246752 exhibited the closest affinity to EU984244 (identified in Taiwan, China, 2008), despite a considerable temporal separation between these isolates. This finding suggests that HBoV1 evolution may not exhibit strict temporal patterns, which is consistent with previous reports describing similar evolutionary dynamics [35].
Genomic sequence alignment revealed that the HBoV isolates that circulated in Beijing during 2023 maintained relatively conserved genetic profiles. All eleven complete genomes were classified as the HBoV1 genotype. The alignment revealed that the sequenced isolates shared 94.7–96.0% nucleotide identity and 94.8–96.1% amino acid identity with the prototype strain NC_007455.1, with only eight nonsynonymous mutations resulting from nucleotide substitutions. In previous studies, the VP1 protein has been shown to have greater genetic diversity. Among the complete sequences, it presented the greatest number of variant sites. We identified seven missense mutations in the VP1 protein. Among them, the N474S substitution was present in all sequenced isolates and represented 95.1% of the complete genomic sequences in GenBank. This phenomenon was also observed in a previous study that was conducted in Kunming [25]which might be attributed to the fact that this stable mutation may increase virus pathogenicity and be related to immune evasion. Moreover, the D180N and G499E substitutions represent previously unreported mutations in the VP1 gene. The N-terminal region of VP1 harbors a unique 129-amino-acid domain (VP1u), demonstrating phospholipase A2 (PLA2) activity that is essential for viral infectivity. Studies have identified four residues (at positions 21, 41, 42, and 63) as crucial for preserving this enzymatic function [36, 37]. Our analysis revealed three amino acid substitutions in the VP1u region, including two (R17K and L40S) within the PLA2 active domain (residues 11–69). These variants were present at frequencies of 8.6% and 8.3%, respectively, in the complete HBoV genomic sequences available in GenBank, potentially affecting viral infectivity through modulation of the host cell entry mechanism.
For the high-frequency mutation N474S, we performed structural predictions to assess its potential impact on the VP1 conformation. The analysis revealed that this substitution alters both the hydrogen bonding network and the interacting amino acid residues within its 5 Å radius. Hydrogen bonds represent a class of intermolecular interactions that are crucial for stabilizing secondary protein structures. These intermolecular forces between amino acid residues promote polypeptide chain folding and maintain conformational integrity, thereby directly affecting protein function [38]. Extensive research has shown that modifications to viral protein hydrogen-bonding networks can substantially influence protein‒host–cell interactions, with significant consequences for viral pathogenicity and infectivity [39, 40]. The N474S mutation may alter VP1 structural stability, thereby potentially increasing viral pathogenicity and accounting for its persistent prevalence in circulating isolates. However, it is important to note that the complete VP1 structure remains undetermined. The VP1 structural model in this study was predicted via AlphaFold3, which may not accurately reflect the native protein conformation. Experimental validation through cryo-electron microscopy or other high-resolution structural techniques will be needed for definitive characterization.
We performed entropy analysis on the three viral proteins to quantify the sequence variability. The results aligned with the observed amino acid substitution patterns: NS1 presented the highest evolutionary conservation among the 11 sequenced isolates, followed by NP1. Conversely, VP1 displayed greater sequence variability, with higher entropy values indicating potential conformational diversity.
Viruses are subject to evolutionary selective pressures that favor the survival and replication of variants with enhanced environmental adaptability. These pressures progressively influence the genetic architecture and phenotypic characteristics of viral populations, thereby facilitating evolutionary adaptation. On the basis of their functional consequences, selective pressures can be categorized into three distinct types: positive selection (directional selection), negative selection (purifying selection), and neutral selection. Our analysis identified one NSS in both the NP1 and VP1 proteins, suggesting that evolutionary constraints are imposed by purifying selection. These observations are consistent with previous reports documenting multiple NSSs in waterfowl parvovirus (Parvoviridae family), which similarly demonstrated strong purifying selection pressures [41]. Mutations at NSSs likely impose evolutionary constraints on viral isolates, as purifying selection typically eliminates variants with alterations at these conserved positions, thereby preserving amino acid sequence integrity and protein structural stability. Although the structural and functional consequences of mutations at these specific sites (86 and 474) remain uncharacterized, our findings establish an important foundation for future mechanistic studies.
Posttranslational modifications (PTMs) substantially modulate protein structures and functions. Among these modifications, glycosylation represents a critical PTM that plays a fundamental role in regulating protein folding, antigenic properties, and biological activity [42]. N-glycosylation does not cause major conformational alterations in proteins but diminishes molecular dynamics while improving structural stability [43]. O-glycosylation plays a crucial role in maintaining the three-dimensional conformation and stability of proteins [44]. Research has shown that glycosylation occurs in various viruses during infection and plays a crucial role in regulating virus‒host interactions. Prominent examples include the human immunodeficiency virus (HIV), influenza A virus (IAV), and SARS-CoV-2, in which viral glycosylation patterns critically regulate both infectivity and immune evasion mechanisms [45, 46]. The characterization of glycosylation sites in HBoV currently represents an understudied research area. In this study, we conducted systematic predictions of potential glycosylation sites across all sequenced isolates to bridge this knowledge gap. Analysis of the 11 sequenced isolates revealed that four contained an acquired N-glycosylation site and concomitant loss of an O-glycosylation site resulting from the NP1 S79N substitution. This modification may induce structural alterations through changes in glycosylation patterns. Notably, all of the sites were identified by in silico prediction and its relevance requires experimental verification.
In summary, our study provides a multilevel genomic characterization of the HBoV isolates circulating in Beijing during 2023, including nucleotide, amino acid, and protein structural analyses. These findings contribute to the global understanding of the genetic diversity of HBoV while offering valuable insights for future investigations into the structure‒function relationships of the VP1 capsid protein. A thorough understanding of viral protein evolutionary and structural characteristics will facilitate the rational design of targeted antiviral strategies and vaccine development.