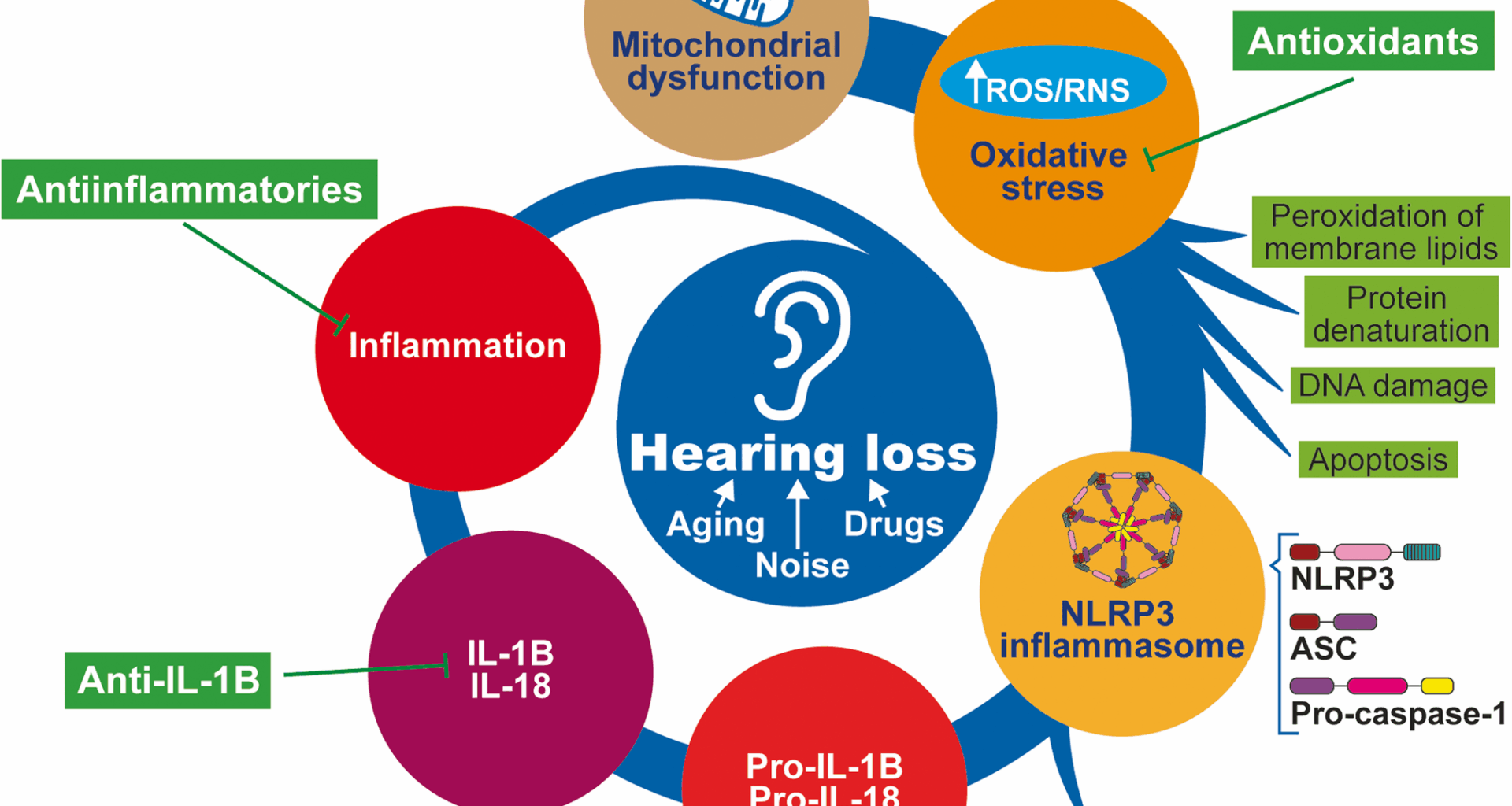
Sensorineural hearing loss (SNHL) affects approximately 1.5 billion people globally. Approximately 430 million of these patients require rehabilitation services for disabling hearing loss [12]. SNHL is caused mainly by dysfunction of the inner ear due to aging, exposure to ototoxic drugs or noise, or mutations in nuclear or mitochondrial genes. However, it is idiopathic in some patients. Although the inner ear was previously thought of as an immune-privileged organ, inflammation—along with oxidative stress—is considered a central pathogenic mechanism of hearing loss [13, 14].
The activation of the NLRP3 inflammasome constitutes a physiological host defense response to danger signals. However, dysregulated inflammasome activity can lead to excessive inflammation, causing substantial damage, especially in those tissues, such as the cochlea, with scarce regenerative capacity. There is increasing evidence that inflammasome activation is associated with hearing loss. Thus, gain-of-function NLRP3 mutations in autoinflammatory diseases are commonly associated with hearing loss. Furthermore, the increase in the cochlear uptake of gadolinium, measured as an increased intensity of the MRI signal, indicates barrier leakage and is normalized by anakinra therapy. The induction of the NLRP3 inflammasome has also been confirmed in experimental models of noise-induced hearing loss [15], ototoxicity induced by aminoglycoside antibiotics [16] or platin-derived chemotherapeutics [11]. In addition, hearing loss can occur due to cytomegalovirus infection during pregnancy [17].
Hearing loss in cryopyrin-associated periodic syndrome
Abnormal hyperactivation of the inflammasome and excessive production of IL-1B are the causes of a spectrum of autosomal dominant systemic autoinflammatory diseases called cryopyrin-associated periodic syndrome (CAPS), which include (in order of severity) familial cold autoinflammatory syndrome (FCAS), Muckle–Wells syndrome (MWS), and chronic infantile neurological, cutaneous and articular (CINCA) syndrome. These diseases have an estimated world prevalence of 2.7–5.5 per 1 million people [18] and are considered ultrarare diseases (ORPHA:208650). However, considering that many patients are diagnosed very late or not at all, its prevalence is likely greater.
The typical inflammatory symptoms observed in CAPS include fever, headache or fatigue, and local symptoms in the skin, joints, muscles, eyes, cochlea and central nervous system [19]. The correct diagnosis and immediate initiation of therapy with IL-1B inhibitors are mandatory in most patients to achieve a reversal of daily symptoms and prevent possible life-threatening sequelae [18].
FCAS was first described in 1940 in a five-generation family exhibiting recurrent episodes of urticarial-like rash, limb pain and fever following cold exposure [20]. Over the next few decades, similar cases with “cold hypersensitivity” were reported and finally classified as FCAS to differentiate this inherited disorder from the more common acquired cold urticaria. In addition to urticaria rash and a burning sensation triggered by exposure to cold, fever, malaise, conjunctivitis, abdominal discomfort and polyarthralgias are very common, whereas amyloidosis and deafness are uncommon or absent.
MWS presents similar symptoms (fever, rash, arthralgia, conjunctivitis, amyloidosis) and sensorineural deafness. The first report of the auditory phenotype in MWS was in 2012, when a single-center MWS cohort (19 patients aged 3–72 years, belonging to four families with three different mutations in NLRP3) was explored with pure tone audiograms, vestibular testing, and tinnitus questionnaires: 89% of them presented bilateral SNHL, which started at high frequencies and led to profound deafness in the most severe cases, and nearly half of the adults reported intermittent or permanent tinnitus [21]. Another study with additional cohorts of MWS patients reported a high percentage (67–92%) of hearing loss [22,23,24].
CINCA, also called neonatal-onset multisystem inflammatory disease (NOMID), was identified in 1987 [25] and represents the most severe phenotype of CAPS, with very early-onset skin rash, arthropathy and severe central nervous system symptoms, including chronic aseptic meningitis, which may lead to brain atrophy and severe intellectual disability [26]. Hearing loss is also a common symptom that occurs within the first years of life. Early anti-IL1B treatment is the standard therapy, reducing the risk of developing major complications.
CAPS is caused by single heterozygous germline or somatic gain-of-function mutations in the human NLRP3 gene. The Infevers database (available at https://infevers.umai-montpellier.fr/, accessed June 17, 2025) currently lists 303 sequence variants of the NLRP3 gene associated with autoinflammatory diseases, mostly substitutions affecting exon 4 (formerly named exon 3) [27], with pathogenic mutations typically located in the NACHT domain of the NLRP3 protein [28, 29]. Among them, 19 variants were specifically associated with CAPS (5 likely pathogenic, 3 variants of uncertain significance).
Genotype‒phenotype correlations are important for identifying predictive disease severity markers. Similarly, the Eurofever Registry analyzed 136 CAPS patients carrying NLRP3 variants and concluded that skin rash, musculoskeletal involvement and fever were the most prevalent features, with neurological symptoms and hearing loss present in 40% and 42% of the patients, respectively [30]. Heterozygous germline mutations were found in 98% of the patients, and only 3 patients were mutation-negative despite complete NLRP3 gene screening. Thirty-one different NLRP3 gene mutations were detected, with 7 accounting for 78% of the patients, and 24 rare variants in 21% of the patients were significantly associated with early disease onset (5, 28, 29].
Although CAPS patients frequently suffer from sensorineural hearing loss, it remains unclear whether NLRP3 mutation is the primary cause of cochlear autoinflammation, which may be the sole manifestation in some CAPS rare cases (DFNA34), or if systemic inflammation contributes to the development of progressive hearing loss. This fact may have an impact on treatment decisions. Notably, there is a window of opportunity to treat patients with anti-IL-1B, and younger patients are most likely to respond. Consequently, it is important to know the characteristics of CAPS for the early diagnosis of associated hearing loss, and mutation analysis of NLRP3 will lead to a definite diagnosis [31].
In addition to CAPS, classical autoinflammatory diseases are characterized by apparently unprovoked inflammation without high-titer autoantibodies or antigen-specific T cells. These manifestations usually include neurological manifestations, such as meningitis, hearing loss, and other nonneurological manifestations. Among the genes involved in these diseases are those encoding MEFV (Mediterranean fever), TNFR (TNF receptor-associated periodic syndrome) or MKV (hyperimmunoglobulinemia syndrome). NLRP3 mutations have also been identified. Thus, Salsano and coworkers demonstrated a novel NLRP3 mutation (p.I288M) and a previously described MEFV mutation (p.R761H) in a patient with a chronic disease characterized by meningitis, osteomyelitis, leukoencephalopathy and progressive hearing loss, along with increased inflammatory markers. Patients respond to tocilizumab (an anti-IL-6 receptor monoclonal antibody) but not to anakinra (a recombinant IL-1R antagonist); therefore, IL-6 hypersecretion is the likely pathogenic mechanism [32].
Nonsyndromic genetic hearing loss
NLRP3 gene mutations are the cause of autosomal dominant autoinflammatory disorders, mostly CAPS, which include (syndromic) hearing loss. However, hearing loss has also been found to be the sole manifestation of these diseases, leading to a misdiagnosis of nonsyndromic deafness [33].
A variety of targeted NGS panels have been developed in recent years for genetic screening of nonsyndromic deafness, but they do not usually include typical syndromic deafness genes, such as NLRP3. Thus, Chen and colleagues conducted genetic screening via targeted next-generation sequencing (NGS) panels in a family with dominant inheritance initially diagnosed with nonsyndromic deafness. No pathogenic variants were found in any of the 72 known genes associated with nonsyndromic hearing loss. However, subsequent whole-exome sequencing identified a heterozygous p.E313K variant in the NLRP3 gene. Follow-up clinical evaluation revealed that 6 out of 9 affected family members presented subtle inflammatory signs that had previously gone unnoticed [8].
Similarly, Nakanishi et al. identified a missense mutation, p.Arg918Gln, of the NLRP3 gene associated with autosomal-dominant nonsyndromic SNHL in two unrelated families [5]. The affected subjects presented an atypical CAPS phenotype, with the sole symptom being a bilateral slowly progressive SNHL with an onset in the late 2nd to 4th decade of life that initially affects high frequencies, which can be improved or stabilized by anti-IL-1 therapy [31, 34].
In a recent study, 110 families with autosomal dominant hearing loss were tested with a custom panel of 237 hearing loss genes, and the NLRP3 c.1872 C >G, p.Ser624Arg mutation was identified in one family [35]. ELISA and bioluminescence assays in peripheral blood mononuclear cells from these patients revealed that this novel gain-of-function mutation led to increased activity of caspase-1 and subsequent oversecretion of proinflammatory IL-1B [35]. Clinical reanalysis of the affected individuals, together with serological evidence of inflammation and pathological cochlear enhancement on magnetic resonance images, guided the diagnosis of atypical NLRP3 autoinflammatory disorder. In summary, genetic analysis in patients with nonsyndromic hearing loss should include genes causing these atypical forms to allow timely and effective treatment with IL-1 receptor antagonists.
Nonhereditary congenital hearing loss
Congenital cytomegalovirus infection. It is the most common fetal viral infection and the leading nongenetic cause of SNHL in children, contributing to 25% of the cases under 4 years of age [36]. Cytomegalovirus infection induces a direct cytopathic effect in spiral ganglion neurons and a cochlear inflammatory response. A study with an experimental model of cytomegalovirus infection-associated hearing loss in newborn mice established that cytomegalovirus induced inflammasome-associated factors in spiral ganglion neurons and increased the content of reactive oxygen species [17]. More recently, cytomegalovirus has been shown to induce spiral ganglion neuron (SGN) death via both apoptosis and pyroptosis, with simultaneous activation of the p53/JNK and NLRP3/caspase-1 signaling pathways, respectively, due to the activity of the mixed lineage kinase family (MLK1/2/3), and the MLK inhibitor URMC-099 can prevent cytomegalovirus-induced SGN death and hearing loss [9].
Bilirubin ototoxicity. An increase in bilirubin levels in newborns can cause toxic effects on the auditory system, leading to hearing loss. Unconjugated bilirubin (UCB) can activate inflammatory mediators such as IL-18 and TNF, although the mechanism at the molecular and cellular levels remains unclear. Ex vivo organotypic cochlear cultures exposed to UCB presented demyelinated nerve fibers and a decreased size of spiral ganglion neurons, along with increased levels of NLRP3, cleaved caspase-1 and GSDMD. In addition, the application of pyroptosis inhibitors reduces the levels of the aforementioned proteins, ASC and IL-18, suggesting that the NLRP3 signaling pathway could be involved in UCB-induced ototoxicity [37].
Drug ototoxicity
Cisplatin-induced deafness. Hearing loss is a serious secondary effect observed after antitumoral treatment with cisplatin, affecting 40–80% of adults and over 50% of children treated with this drug [38]. Cisplatin cytotoxicity is generally mediated through DNA crosslinking and reactive oxygen species production. The high susceptibility of the cochlea to cisplatin damage is due, in part, to long-term retention of cisplatin in the stria vascularis, where it induces an inflammatory response and marginal cell damage [39, 40].
A recent in vitro study confirmed that in response to cisplatin, marginal cells exhibit increased expression of NLRP3, caspase-1, IL-1B, and GSDMD, along with the formation of cell membrane pores. This situation was reversed by downregulation of NLRP3 by small interfering RNA, suggesting that NLRP3 inflammasome activation may mediate cisplatin-induced marginal cell inflammation and pyroptosis in the cochlear stria vascularis [11].
Additional mechanisms linking cisplatin ototoxicity to NLRP3 inflammasome activation have been described. First, cisplatin significantly decreased the levels of POU4F3, a transcription factor encoded by a well-known dominant nonsyndromic deafness pathogenic gene (DFNA15). Recently, Pou4f3 mutations were shown to promote cochlear hair cell pyroptosis by activating the NLRP3/caspase-3/GSDME pathway. Therefore, Pou4f3 knockdown via shRNA can be combined with cisplatin treatment to induce pyroptosis in cochlear hair cells through the NLRP3/caspase-3/GSDME pathway [41]. Second, a retrospective cohort study with patients receiving cisplatin chemotherapy with or without concomitant antidepressive treatment revealed that the risk of ototoxicity was lower in the group treated with the selective serotonin reuptake inhibitors fluoxetine or fluvoxamine, which have been shown to inhibit the NLRP3 inflammasome [42]. In summary, the NLRP3 inflammasome plays a pivotal role in mediating cisplatin-induced ototoxicity through different mechanisms.
Aminoglycoside ototoxicity. Several antibiotics can induce hearing loss in children and adults, and the accumulation of oxygen radicals and inflammation in the inner ear are considered central pathological mechanisms. A recent study investigating whether the NLRP3 inflammasome is involved in aminoglycoside-related hearing loss revealed that mice treated with kanamycin plus furosemide presented increased levels of NLRP3 and increased levels of activated caspase-1, IL-1B, IL-18, and GSDMD-N and that oridonin treatment reversed this situation [16]. Furthermore, another study demonstrated that pharmacological inhibition of NLRP3 via MCC950, as well as genetic deletion of NLRP3, significantly protected against SGN degeneration in patients with aminoglycoside-induced hearing loss [43].
Noise-induced hearing loss (NIHL)
Exposure to acute high-intensity noise can severely damage cochlear structures and induce the activation of DAMPs, which are recognized by innate immune receptors, triggering an inflammatory response [44]. Recent studies have confirmed increases in the levels of NLRP3, cleaved caspase-1, IL-1B, and IL-18 in the cochleae of minipigs and in mice exposed to 120 dB SPL noise, suggesting that the activation of the NLRP3 inflammasome constitutes a central pathogenic mechanism in NIHL [15, 45]. Moreover, the use of anakinra or oridonin has been proven to be effective in protecting mice from NIHL by facilitating inflammasome complex assembly [45, 46].
Chronic exposure to moderate levels of noise also affects the inner ear and reorganizes central auditory pathways, although the role of NLRP3 remains to be elucidated. In a study from Feng and collaborators, C57BL/6J mice were exposed to long-term 70 dB SPL white noise, aggravating the concomitant age-related hearing impairment typical of this strain [47]. They reported that cochlear ribbon synapses were the primary site of inner ear injury caused by chronic noise exposure. These authors confirmed by western blotting the presence of a significant increase in the levels of NLRP3, caspase-1 and IL-1B in P3 mouse cochlear explants exposed to NMDA and kainate to mimic noise-induced excitotoxic damage. These results indicate that NLRP3 inflammasome is an important mechanisms underlying auditory nerve fiber damage after noise [47].
Age-related hearing loss (ARHL)
ARHL, or presbyacusis, is a progressive loss of hearing sensitivity predominantly associated with hair cell and SGN degeneration in the inner ear. Oxidative stress and a chronic low-level inflammatory response are frequently found in aging cochleae. Although inflammasomes are likely responsible for the accumulation of reactive species in immune cells, whether they are involved in the development of ARHL is still unknown. A study in mice demonstrated via RT‒qPCR, western blotting and ELISA that the levels of activated NLRP3, caspase 1, IL-1B and IL-18 were significantly greater in the inner ears of aged mice than in those of young mice [48].
Meniere’s disease
Meniere’s disease is an inner ear disorder characterized by severe vertigo episodes and hearing loss. The causes and precise pathological mechanisms remain undefined, although alterations in immune responses have been proposed. Recently, downregulation of serum/glucocorticoid-inducible kinase 1 (SGK1) was shown to be associated with activation of the NLRP3 inflammasome in vestibular resident macrophage-like cells from Meniere’s disease patients [49]. Moreover, Sgk−/− mice that received LPS presented severe audiovestibular symptoms, increased inflammasome activation and endolymphatic hydrops, which were ameliorated by blocking NLRP3. Pharmacological inhibition of SGK 1 also increases disease severity in vivo. SGK1 phosphorylates the NLRP3 PYD domain, which acts as a physiological inhibitor of NLRP3 inflammasome activation to maintain inner ear immune homeostasis. SGK1 depletion enhances the NLRP3 inflammasome and IL-1B production, potentially leading to damage to inner ear hair cells and the vestibular nerve. Thus, SGK1 inhibition could offer an alternative to current treatments based on corticosteroid administration [50].
Vestibular Schwannoma
Vestibular schwannomas arise from neoplastic Schwann cells of the vestibular nerve and constitute the fourth most common type of intracranial tumor, often causing SNHL and tinnitus [51]. There was no correlation between tumor size and the grade of hearing loss, suggesting that vestibular schwannoma-associated SNHL is due not only to mechanical compression of the auditory nerve but also to differences in the intrinsic biology of these tumors. Previous research has reported an abnormal upregulation of inflammatory pathways in these tumors and a correlation between poor hearing and a robust inflammatory response in vestibular schwannoma patients. A meta-analysis of a large vestibular schwannoma microarray dataset by Sagers and collaborators identified the NLRP3 inflammasome as a candidate, which was further validated in human vestibular schwannoma tissue via RT‒qPCR and immunohistochemistry [52]. In addition, the authors reported an association between the overexpression of NLRP3 inflammasome components in vestibular schwannoma and a high degree of hearing loss. Therefore, the inhibition of the NLRP3 inflammasome in vestibular schwannoma could contribute to preserving hearing.