Clinical phenotypes of two patients with apparent homozygous mutations
Patient 1: A 4-month-old boy from a non-consanguineous Chinese parent with a history of recurrent diarrhea and oral ulcers during the neonatal period presented to the gastroenterology department. His 3-year-old sibling sister was healthy. He presented hematochezia at one month, perianal ulceration with fever at two months, and perianal abscess at three months. Exclusive enteral nutrition with an amino-acid-based formula and anti-infective therapy were not effective. He also had anemia, hypoalbuminemia and leukocytosis, and elevated C-reactive protein. Lower endoscopy demonstrated ulcerations in the sigmoid and descending colon, polypoid lesions in the descending colon and mild stenosis of descending colon. Rectal biopsy pathology suggested high proliferation of mucosal lymphoid tissue (Table 1).
Table 1 Clinical phenotypes of two patients with apparent homozygosity in IL10RA
Patient 2: A 6-month-old girl from a non-consanguineous Chinese parent with a history of recurrent fever and diarrhea during the neonatal period presented to the gastroenterology department. She also developed rectoperineal and rectovaginal fistulas, umbilical hernia, and oral ulcer. Antibiotics and parenteral nourishment in the local hospital did not alleviate her symptoms. Laboratory tests revealed anemia, leukocytosis, elevated C-reactive protein and erythrocyte sedimentation rate, and hypoalbuminemia. Colonoscopy revealed ulcerations and pseudo-polyps in the rectum and sigmoid colon and stenosis of the sigmoid colon. Pathology of small bowel biopsy suggested a significant number of lymphocytes in the mucosa (Table 1).
Genetic sequencing of two patients with apparent homozygous mutations
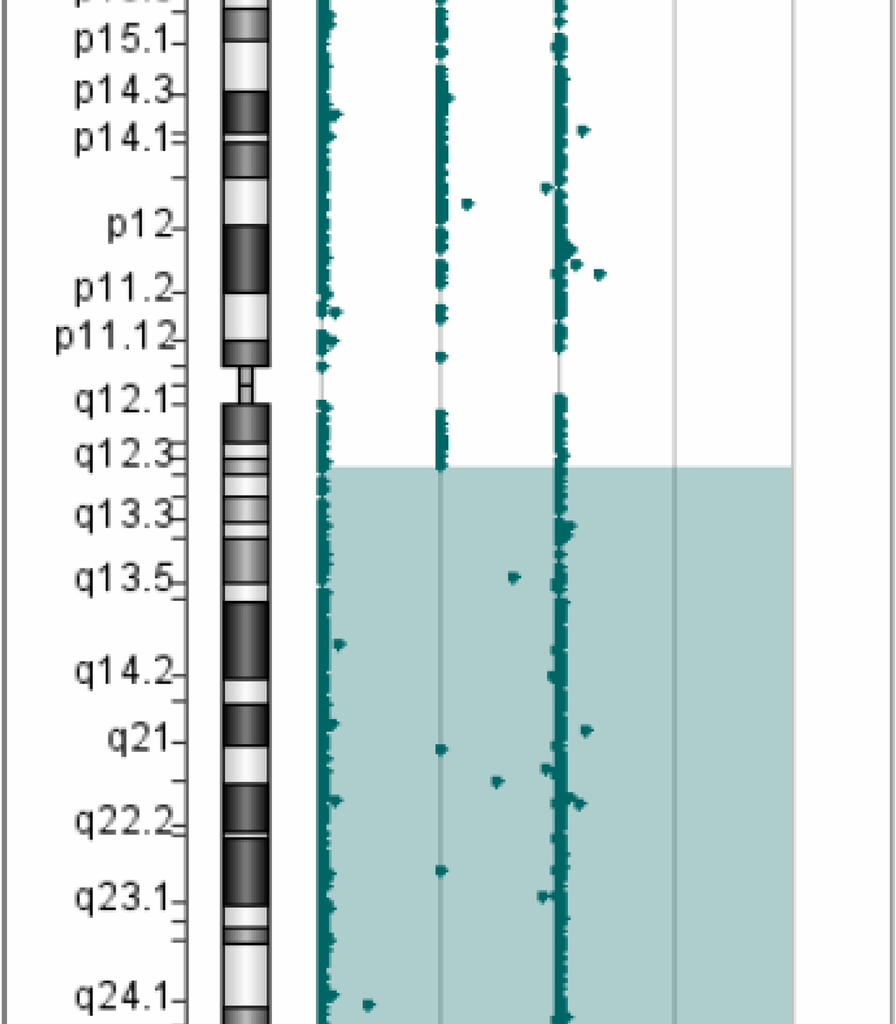
In Patient 1, WES revealed a homozygous mutation in the IL10RA gene (NM_001558.4: exon 3: c.301C > T chr11-117860269 (GRCh37) p.R101W) on the long arm of chromosome 11. The variant was previously reported to be pathogenic (Accession: VCV000039432.8). However, the origin of the variant suggested that the father was heterozygous and the mother had no variant, which was inconsistent with an RA mode of inheritance. Further analysis of the Agilent arrayCGH 4 × 180 K custom chip showed 0 and 2 uncut alleles, and suggested that Patient 1 had a loss of heterozygosity (LOH) in the 72.2 Mb chromosomal segment ranging from 11q12.3 to 11q25, containing IL10RA mapped to the area of 11q23.3 (Fig. 1). Combined pedigree analysis, clinical phenotype, and genetic analysis revealed that the disease could be caused by partial uniparental disomy [upd(11)pat] affecting the expression of imprinted genes, explaining why both variants are from the same paternal line (Fig. 2A).
Fig. 1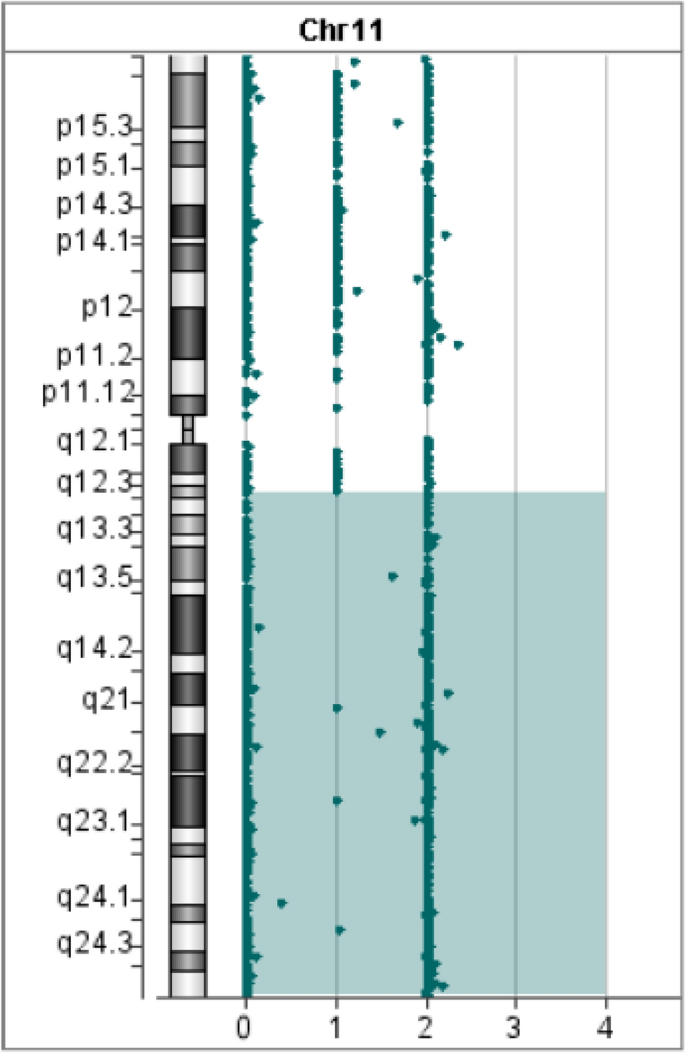
Gene microarray testing of Patient 1. The bottom number is the log2 ratios for the CGH-only microarrays
Fig. 2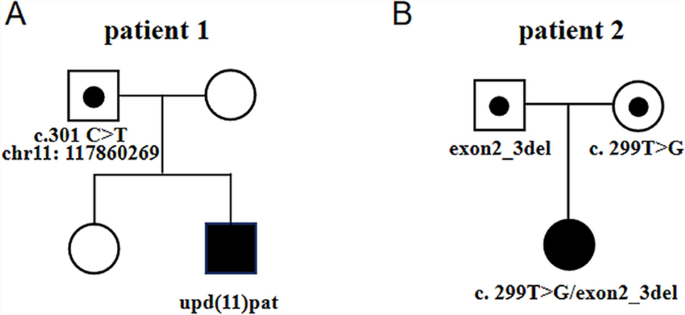
Pedigrees of the two cases. A Pedigree of a Chinese family with autosomal recessive IL10RA-deficient IBD owing to complete paternal uniparental disomy. B Pedigree of a patient with a large fragment loss in exons 2 and 3 at IL10RA. The spot represents the carrying status, while the blue filling represents the disease status
Patient 2 carried the homozygous mutation c.299 T > G in IL10RA, as confirmed by WES analysis. This variant has been reported before and has been proved to be deleterious [4, 15]. Sanger sequencing showed that the mother was heterozygous for IL10RA c.299 T > G, while the variant was not found in her father (Fig. 3). CapCNV analysis of WES data found the copy number variation in the other allele, which was a loss of large fragments spanning exons 2 and 3. Quantitative real-time quantitative polymerase chain reaction (qPCR) showed the loss of exons 2 and 3 in father and patient (Fig. 4). The point mutation and copy number variation are coincidentally located in the same area (exon 3) and were therefore initially incorrectly inferred to be homozygous mutations, whereas a combination of lineage analyses (Fig. 2B) and genetic testing ultimately determined the genotype to be a compound heterozygosity. Multiexon deletion, one type of null variant, is usually regarded as pathogenic variation according to ACMG guideline [4, 14].
Fig. 3
Sanger sequencing of the family of Patient 2
Fig. 4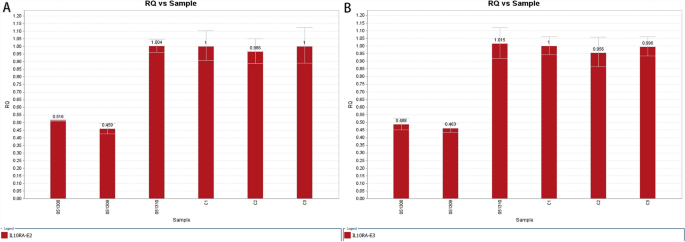
qPCR of exon 2 and exon 3 of IL10RA in the patient 2’s family. RQ is short for ratio relative quantity. ID 051308, ID 051309 and ID 051310 represent patient 1, the father and the mother respectively.C1, C2 and C3 belong to normal healthy people. Figure 1A shows the quantity of exon 2, while Fig. 1B shows that of exon 3. The reference gene is exon 3 of ALB, and relative quantity is calculated by 2−ΔΔCt method
Literature review of patients with large fragment deletion in IL10RA
The review included 15 patients from diverse geographical and ethnic backgrounds: 12 cases from China, 1 from South Korea, 1 Arabic individual, and 1 White individual (Table 2). All parents of the patients were non-consanguineous, with the exception of case 1. 2 patients (case 1 and case 11) exhibited substantial deletions in both alleles of IL10RA. 13 individuals had base mutations in one allele and deletions in a substantial section of the other allele in IL10RA. In the majority of these individuals, the segmental deletion was situated in exon 1.
Table 2 Report on gene deletion of IL10RA
All patients had onset of disease in childhood, with 14/15 patients having onset of disease within one year of age. The primary clinical symptoms included severe diarrhoea and perianal abscesses. 8 cases were documented endoscopically in the literature as exhibiting severe colitis, colorectal ulcers, intestinal stenosis, or intestinal perforation. 5 patients were reported to have experienced bloody stools over the course of the disease, while 7 patients were noted to have anal fistulae. Alongside gastrointestinal symptoms, all patients had extraintestinal signs, including perianal lesions, recurrent fever, mouth ulcers or infections. Among the laboratory parameters, elevated White blood cell (WBC), CRP and abnormalities of lymphocytes and immune function were mentioned in some papers. Case 2 had enterostomy for intestinal perforation, whereas Case 8 received enterostomy due to severe infections and perianal lesions. 4 patients had HSCT, and 1 succumbed to post-transplant sepsis. In conclusion, all patients had an early start of illness, extensive systemic lesions, and poor prognosis, requiring the selection of appropriate treatment.