Permitting
The analysis of human-derived eDNA from field obtained samples and human-related sampling was conducted with University of Florida (UF) Institutional Review Board (IRB-01) ethical approval under project number IRB202201336, with all study participants providing informed consent. One participant provided sand footprint samples, and six participants provided room air samples (pooled room air). Participation was on a voluntary basis, with no compensation received by the participants. Permitting for the collection of our water and sediment samples that were used as outgroups here is as reported in the publications for which those samples were originally collected6,7,8.
Sample collection
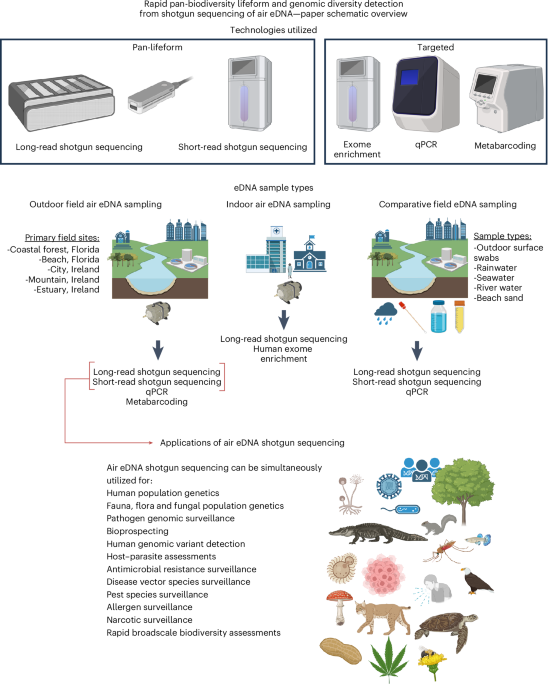
In this study, we generated 78 shotgun sequencing datasets (30 ONT and 48 Illumina), 4 human exome-enriched deep-sequencing datasets (Illumina), 14 vertebrate 12S metabarcoding datasets and assessed 62 samples by qPCR (Supplementary Table 1). Of the shotgun sequenced samples, nine were ONT outdoor active pump sampling, and 25 were Illumina outdoor active pump sampling. Sequenced outdoor samples ranged from 7 h to 35 days of pumping, with 1 week being the standard duration employed.
All laboratory procedures from sampling to final analysis (sequencing) were conducted in a way that minimized DNA contamination from investigators or other sources. This included no contact of investigators/samplers with substrates being sampled (water, sand or air), new nitrile gloves for each sample collection and frequent glove changes throughout sample processing, cleaning of equipment and benchtops with bleach before use, and NFCs being treated identically to genuine field samples throughout all processes from extraction through to sequencing or qPCR. All eDNA samples were extracted in laboratories that do not process other biodiversity DNA samples: a sea turtle laboratory in Florida and a chick/mouse developmental biology laboratory in Ireland.
All outdoor air samples and Irish sand samples were collected for this study. The outdoor air samples were collected by one of three methods: 0.22-µm pore Millipore Sterivex-GP Pressure Filter Units (Merk Millipore, cat no. SVGPL10RC) (1) attached to a vacuum pump (Fisherbrand MaximaDry (115 V, 40 l min−1, cat. no. 1388016) or Fisherbrand MaximaDry (230 V, 20 l min−1, cat. no. 1388015)) and filtered for a specific duration (active pump sampling), (2) left in an environment to passively absorb airborne DNA (passive sampling), or (3) taped to the roof of a vehicle with air passing through the filter while the vehicle drove (car mounted, Ford Explorer) (Supplementary Fig. 1a). The Irish air vacuum pump samples were collected for one extra day compared with the Florida 7-day samples, to partially offset the use of a slightly weaker vacuum pump. The car-mounted samples were not actively vacuum pumped but rather air passed through freely as the vehicle drove (Fig. 2a and Supplementary Fig. 1a). The car-mounted sampling route was a round trip (4 h 20 min of driving) from our research institute to our main campus sequencing facility (short amount of barrier-island driving but predominantly inland habitats that included urban towns, agricultural areas and conifer forests). Air NFCs had filters placed in the same environment (adjacent to sampling filters) for the same duration but with luer-lock caps in place to prevent any airflow through the filter.
Non-Irish seawater and beach sand eDNA samples were collected between 2018 and 2023 and sequenced as part of our human, sea turtle and pathogen research6,53. The soil samples were collected (2022) as part of a bobcat eDNA study8. Indoor air samples from our sea turtle hospital were collected (with ethical approval from the UF IRB, 2022–2023) as part of a human eDNA study (2022 and 2023)7, but the indoor air human exome enrichment sequencing was conducted for this current study. The NFC samples of water and sand were also collected and processed as per the study samples. For the NFC water sampling, 1 litre of MilliQ water (Florida) or 1 litre of Qiagen Nuclease-free water (Ireland, cat. no. 129117) was transported from the laboratory to field sampling locations and stored in a cool box with the environmental samples to monitor for potential contamination during sampling, transportation and processing7. The air and water NFCs were filtered and extracted alongside the other collected air and water samples from each sampling trip and subjected to the same next-generation sequencing or qPCR conditions. Between 300 ml and 1 litre of water samples were filtered (Supplementary Table 1); lower volume samples had been filtered until debris clogging prevented larger volumes being filtered. For low debris samples (for example, February 2024 rainwater) a maximum of 1 litre was filtered, despite filters not becoming clogged. For sand/soil eDNA, a 50-ml tube was filled with sand from each sampling event, with 10 ml of this sand/soil used per individual eDNA extraction6. Human-present (sea turtle rehabilitation hospital) air samples were collected from a 280-square-foot room while participants went about their daily work activities (that is, could enter and exit the room throughout sampling period), with a maximum of the same six participants using the room for a portion of the sampling period7. The room was air conditioned (outside air) and had an external door that was opened and closed and occasionally left open for certain work procedures. Indoor air was also collected from an office-style building on a barrier island (24 h vacuum pumped, 24 h office air May 2023), which had been temporarily closed (no occupants) to facilitate old ceiling tile and roof insulation removal. The NFC samples of air were also collected and processed as per the study samples. For outdoor air sampling, pumps were only started once investigators had left the air filter inlet location, and pumping was stopped before the recovery of the filters.
DNA extraction and sequencing
Before filtration and between every sample, laboratory surfaces, filtration equipment and all standard laboratory equipment involved in the washing and filtration process, as well as the filtration pump itself were disinfected with 70% ethanol (general cleaning and biotic disinfection), and sampling equipment (collection bottles) was disinfected by washing thoroughly with 10% bleach (DNA destruction) and rinsed thoroughly with deionized water.
Outdoor air
The outdoor air samples were collected as described above. Immediately after air sampling, 740 µl of Buffer ATL was added into the filter units, and the samples were either immediately extracted or filters stored at −20 °C in Buffer ATL until extraction. Next, 20 µl proteinase K (from 1 h to 24 h) was added to each filter (after filter thawing if it had been stored frozen) and the 56 °C ATL Buffer and proteinase K incubation was conducted for 1 h. For four samples, the 1-h incubation was reduced to a 10-min incubation to assess the feasibility of reduced extraction times for outdoor air samples.
Sand
For sand samples, 1× concentration Tris and EDTA (TE) buffer (IDTE pH 8.0 1× TE solution) (Integrated DNA Technologies, cat. no. 11-05-01-09) was added to each individual sand sample in individual 50-ml Falcon conical centrifuge tubes, at approximately two times the volume of sand (10 ml of sand and 20 ml of 1× TE buffer). The samples were shaken gently by hand, then set on a rocking platform for 1 h at room temperature. The samples were rested until sand had sunk to the bottom of each tube, then the supernatant was immediately pipetted into a 60-ml sterile BD luer-lock syringe (Fisher Scientific, cat. no. 136898). The samples were then hand filtered using 60-ml BD luer-lock syringes through 0.22-µm Sterivex-GP Pressure Filter Units (Millipore, cat. no. SVGPL10RC) and capped with B.Braun luer-lock caps (Medline, cat. no. BMGTMR2000B). A total of 740 µl Buffer ATL and 60 µl proteinase K from a Qiagen DNeasy Blood and Tissue Kit (Qiagen, cat. no. 69504) were added to each sample, and they were placed in 50-ml Falcon conical centrifuge tubes in a rolling incubator overnight at 56 °C.
River/seawater
The water samples were pumped (by hand, Ireland and Florida 2023, or electronically, Florida before 2023) through 0.22-µm Sterivex-GP Pressure Filter Units (Millipore, cat. no. SVGPL10RC) and capped with B.Braun luer-lock caps (Medline, cat. no. BMGTMR2000B). Hand pumping was with sterile 60-ml BD luer-lock syringes. The electronic pumping was carried out using a GeoTech Peristaltic Pump Series II. A total of 740 µl of Buffer ATL and 60 µl of proteinase K from a Qiagen DNeasy Blood and Tissue Kit were added to each sample. and they were placed in 50-ml Falcon conical centrifuge tubes in a rolling incubator overnight at 56 °C.
Rainwater
Rainwater was collected using sterile 1-litre bottles and sterile funnels (Fisherbrand utility 203 mm funnels, cat. no. 1050010) at the Florida hammock forest site (placed on a balcony in an open canopy area with no tree cover). Rainwater was collected in February, August (category 1 Hurricane Debby) and September 2024 (category 4 Hurricane Helene) 2024. The sampling duration ranged from 19 h and 36 min to 20 h and 15 min, and the sampling was stopped once rain ended. The rainwater volumes ranged from 1 litre to 0.45 litre, depending on rainfall volumes. The collected rainwater was then filtered immediately and eDNA extracted as described above for river water, the only exception being that due to the relative clarity of the sample the 56 °C step was conducted only for 2 h and 30 min. For the rainwater NFCs, 1 litre of MilliQ water in a capped sterile bottle was placed beside the rainwater sampling bottles for the duration of sampling and an equal volume processed identically alongside the rainwater samples.
Indoor air
For indoor air eDNA sampling, room air was passed through 0.22-µm pore Millipore Sterivex-GP Pressure Filter Units (Merk Millipore, cat. no. SVGPL10RC), using a Welch vacuum pump (2019LD-4112), a GeoTech Peristaltic Pump Series II or a Fisherbrand MaximaDry vacuum pump (cat. no. 1388016). The eDNA was extracted6,53 as for the water and sand samples, with the exception that only 20 μl of proteinase K was added per filter, and the 56 °C ATL Buffer and proteinase K incubation was conducted for 1 h.
Non-filter based collection
For the car-mounted (Ford Explorer, Supplementary Fig. 1a) air eDNA collection syringe-only sample (4.3 h car-mounted syringe, May 2023), the car-mounted sterile syringe (BD luer-lock syringe, Fisher Scientific, cat. no. 136898) was capped (leur lock cap) and the walls of the syringe washed (pipetting and hand rotation) with 2,960 µl of ATL Buffer. The syringe was then uncapped, and a sterile plunger was used to pass the liquid into a sterile 15-ml tube. A total of 80 μl of proteinase K was then added to the ATL Buffer, and the sample was incubated for 1 h at 56 °C. The Irish windowpane swab (Wicklow Mountain, May 2023) was collected from a windowpane (outdoor facing side, exposed to the elements) at the air sampling site (Supplementary Fig. 1a). The windowpane had been washed 1 year prior (with water and detergent), so it had been accumulating DNA for 1 year. A sterile swab was dipped in ATL Buffer and used to swab a small section of the windowpane. For the Florida swabs, a variety of surfaces at the hammock forest air sampling site were similarly swabbed within 10 min of each other: windowpane, varnished wooden plank, metal fence post and a Palmetto palm (Sabal palmetto) leaf. For all the swab samples, an area of approximately 3 cm2 was swabbed for 20 s. The swabs were immediately placed in a sterile 50-ml Falcon conical centrifuge tubes containing 500 µl of ATL Buffer and stored at −20 °C. For the swab NFCs, a swab was placed directly into a sterile 50-ml tube containing ATL, without being used to swab any surface. After thawing, 60 μl of proteinase K was added to the ATL Buffer, and the sample was incubated for 1 h at 56 °C.
For all three filtered sample types (sand, water and air), after the 56 °C incubation, the solutions of Buffer ATL (post water or air filtration or post sand washing and filtration), proteinase K and eDNA were transferred from the Sterivex-GP Pressure Filter Units to 2-ml microcentrifuge tubes using 10 ml of BD Slip Tip Sterile Syringes (Fisher Scientific, cat. no. 14823434). The DNA was then isolated using a modified Qiagen DNeasy Blood and Tissue Kit protocol6,53,54. Following incubation, equal volume 800 µl (sand and water) AL Buffer and 800 µl (sand and water) ice-cold ethanol were added to each sample, and they were vortexed vigorously and microcentrifuged after each addition. For air samples, 500 µl of each solution was used (as a lower volume of ATL solution is recovered from the initially dry Sterivex filters). For swab samples, 500 µl of each solution was used (directly into the 50-ml tube). Each sample was extracted according to the remainder of the kit protocol, with the only alteration being that DNA was eluted with 70 µl of AE Buffer, incubated at 70 °C before addition to spin column and incubated on the column at room temperature for 7 min before final centrifugation. The DNA concentration was measured on a ThermoScientific Nanodrop 2000 Spectrophotometer (Fisher Scientific), and the samples were stored at −20 °C until shotgun sequencing or qPCR.
Library preparation and Florida Illumina shotgun sequencing was conducted at the UF’s Interdisciplinary Center for Biotechnology Research (ICBR) Core Facilities on an Illumina NovaSeq 6000 (from 2022 to July 2023, 2× 150-bp PE sequencing on an S4 flow cell) or NovaSeq Series X Plus (from October 2023 to 2024, 2× 150-bp PE sequencing on an 25B flow cell) (see Supplementary Table 1 for the sequencing machine utilized for each sample). Indoor air human Illumina exome capture libraries were also constructed and sequenced at the UF ICBR Gene Expression and NextGen Sequencing Cores. Four indoor air eDNA samples (two air eDNA samples and two NFCs) were used for Illumina DNA Prep with Enrichment, Exome Panel (cat. no. 20020183, captures 45 Mb of exonic content) exome capture analysis, according to the manufacturer’s instructions. Exome sequencing was conducted on an Illumina NovaSeq 6000 S4 flow cell for 2× 150-bp cycles (with NFCs expected to return fewer reads due to a lack of human eDNA) (Supplementary Table 1). The Irish samples were shotgun sequenced commercially at BMKGENE, with Illumina libraries sequenced on a NovaSeq 6000 (2023 sequencing, 2× 150-bp PE, eight PCR amplification cycles during library preparation) or a NovaSeq Series X Plus (2024 sequencing, 2× 150-bp PE sequencing on an 25B flow cell) and ONT libraries sequenced on a PromethION 48. All Florida Oxford nanopore samples were sequenced on a MinION in the Duffy lab at the UF’s Whitney Laboratory for Marine Bioscience. MinION libraries were prepared according to manufacturer’s instructions, using the following kits: ONT Ligation Sequencing Kit (SQK-LSK110, cat. no. 76487-106 or SQK-LSK114), NEBNext Companion Module for ONT Ligation Sequencing Kit (cat. no. E7180S), ONT Flow Cell Wash Kit (EXP-WSH004, cat. no. 76487-116) and sequenced on ONT Minion Flow Cells (cat. no. 76487-106). The samples sequenced up to May 2023 were with v10 chemistry (Ligation Sequencing Kit, SQK-LSK110) and R9.4.1 flow cells, while samples sequenced after May 2023 were sequenced with v14 chemistry (SQK-LSK114) and R10.4.1 flow cells (FLO-MIN114.8) (Supplementary Table 1). The MinION samples sequenced after December 2023 used the new ONT light shield (Supplementary Table 1 and see Supplementary Fig. 7c for indicative read length distributions). Throughout the project, the latest sequencing approaches (chemistries/machines) were applied as they became available, to represent the most current technology and to avail of the associated cost savings.
The vertebrate metabarcoding was conducted according to Lynggaard et al.55 using the 12SV05 vertebrate mitochondrial metabarcoding primer set 12SV05 forward: TTAGATACCCCACTATGC and 12SV05 reverse: TAGAACAGGCTCCTCTAG14,55,56. Input DNA (air eDNA or NFC) was amplified on a Bio-Rad C1000 Touch Thermal Cycler with AmpliTaq Gold DNA Polymerase (ThermoFisher, cat. no. N8080241) for the following cycles: 1 cycle of 95 °C for 10 min, then 45 cycles of 94 °C for 30 s, 51 °C for 30 s and 72 °C for 1 min. The PCR products were then purified using a QIAquick PCR Purification Kit (Qiagen, cat. no. 28104), according to the manufacturer’s instructions. The PCR products were used to construct Illumina sequencing libraries at the UF ICBR Gene Expression core facility, and the libraries were paired-end sequenced (2× 150 bp) at the UF ICBR Next-Generation Sequencing core facility, on an Illumina NovaSeq Series X Plus (10B flow cell). Equal pooling ratios utilized 4.42% of a lane for these three 14 eDNA samples and 65 other metabarcoding samples (not for this study). Each of the 14 samples had 0.03–0.09% of the final lane output (or 466,890–1,285,665 reads).
Bioinformatic analysis
All bioinformatic tools were utilized using default parameters, unless otherwise stated.
Non-deuterostome metagenomics
Nanopore fast-based calling with Dorado was utilized. CZ ID37,38, a free, cloud-based metagenomics platform (https://czid.org/) was utilized for biodiversity bioinformatic analysis (ONT and Illumina). Approximately half of the ONT samples analysed were uploaded to the CZ ID bioinformatics pipeline through a mobile/cellular internet connection (Huawei B525s-23a domestic cellular connection router with a Three Ireland mobile network SIM card, over an older 4G network) from our Irish Croghan Mountain field site. The CZ ID platform queries genetic information from all species, excluding deuterostomes. The CZ ID nanopore metagenomic pipeline was utilized for ONT samples. The CZ ID Illumina metagenomic pipeline was utilized for short-read samples. As the CZ ID platform was primarily developed for microbial metagenomics, it does not query deuterostome sequences, as these are identified as host species, and it actively subtracts host reads such as human reads. Moreover, for samples with over 1 million reads, the pipeline actively subsamples only 1 million reads per analysis. OTUs from the CZ ID data were identified twice: those identified with a single sequence and those identified by at least two. The heat maps were generated using the CZ ID heatmap tool. The CZ ID platform38 was also used to determine the presence of AMR genes (CZ ID AMR Pipeline v1.3.1) in Illumina shotgun sequenced samples (Supplementary Table 1).
Metazoan metagenomics (including deuterostomes)
To conduct metazoan long-read and short-read metagenomics, including deuterostomes, the CZ ID nanopore and CZ ID Illumina metagenomic pipelines were recreated, respectively, in a high-performance computing (HPC) environment using Bash and Python, without host filtering, without subsampling to 1 million reads and aligning to all metazoan species (NCBI Blast). To classify metagenomic reads (both long and short) based on taxonomy, DIAMOND v2.1.7 (ref. 57) was utilized for its inherent high-speed alignment, sensitivity and the translation of nucleotide reads for protein alignments capabilities. First, metagenomic reads from eDNA samples were preprocessed, for adaptor trimming and quality control using Trim galore v0.6.7 (Trim Galore! (https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/)) for Illumina or Porechop v0.2.4 (ref. 58) for ONT reads. The two-step DIAMOND process involved database preparation and read alignment. A reference database was constructed using the non-redundant protein sequences from NCBI. Taxonomic information was integrated using DIAMOND’s ‘makedb’ option with the following input files: a protein accession to taxonomy ID mapping file prot.accession2taxid (https://ftp.ncbi.nih.gov/pub/taxonomy/accession2taxid/), a taxonomy nodes file nodes.dmp (https://ftp.ncbi.nih.gov/pub/taxonomy/new_taxdump/) and a taxonomy names file names.dmp (https://ftp.ncbi.nih.gov/pub/taxonomy/new_taxdump/). For both long-read and short-read samples, the processed reads were aligned to the prepared database using DIAMOND’s blastx mode with ‘very-sensitive’ option, ensuring comprehensive taxonomic classification as per the most stringent options of the package. This step translated nucleotide sequences into protein sequences, aligning them against the database. The alignment output format was specified so as to include detailed taxonomic information for each read. Only the best hit for each read was retained using the option ‘max-target-seqs 1’. The results were parsed with in-house python script to aggregate reads on the basis of the NCBI taxonomy structure for different phyla of protostomes and deuterostomes. This reduced version of the CZ ID pipeline was used to overcome the read subset limitations and the exclusive identification of eukaryotes. Therefore, the contig generation step of the pipeline was not implemented, as it is not feasible to reconstruct species-specific contigs for eukaryotes due to the large genome sizes, the pooled nature and the unequal representation of species in eDNA NGS data. The metazoan metagenomics bioinformatics pipeline (MetaBioTax v1.0) has been deposited in GitHub (https://github.com/nousiaso/MetaBioTax, https://gitfront.io/r/nousiaso/QZwYQLPm9dJQ/MetaBioTax/).
Direct reference genome and mitogenome alignments
The Galaxy platform58,59 (https://usegalaxy.eu/) was utilized for bioinformatic analysis with selected reference genomes (Supplementary Table 2), with NanoGalaxy also utilized for nanopore sequenced data60. All samples were checked for quality (FastQC, v0.73 (ref. 61)), adaptors and low-quality reads trimmed (https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/) v0.6.7 for Illumina data; Porechop v0.2.4 (ref. 62) for ONT sequence data), and high-quality reads aligned to the human reference genome (T2T-CHM13v1.1 (ref. 63)) using Bowtie2, v2.4.2 (ref. 64) for Illumina or minimap2, v2.24 (ref. 65) for ONT. Paired-read alignments were conducted for Illumina data and single read alignments for ONT data. The trimmed air eDNA nanopore data were also aligned (minimap2, v2.24 (ref. 65)) to reference genomes (Supplementary Table 2). Human mitochondrial haplogroups were classified from Illumina data using Haplogrep (V3.2.1)66 with the Phylotree 17.2 (ref. 67) and the Kulczynski distance with only sequences passing quality control (>90%) retained. Human mitochondrial haplogroup charts were produced in RStudio68, using the webr package v0.1.5 (https://github.com/cardiomoon/webr). Moth (C. sasakki) and Loblolly pine (Pinus taeda) mitochondrial and chloroplast sequences (ONT data) were concatenated in Geneious, and the genome coverage plots were created using the ggplot package v3.4.0 (ref. 69). Illumina mitochondrial sequences of the bobcat (L. rufus) and the golden silk orb weaver spider (T. clavipes) were aligned to Genbank references (Supplementary Table 3) using the Geneious Mapper with maximum sensitivity and slow mapping options selected (GeneiousPrime 2023.0, www.geneious.com/prime). Jukes–Cantor maximum-likelihood trees were similarly created in GeneiousPrime with automatic sequence determination, global alignments with free end gaps and Identity (1.0/0.0) Cost Matrix options selected, using cougar (Puma concolor) and Joro spider (Trichonephila clavata) as outgroups for the bobcat and spider trees, respectively. UpSet plots and Venn diagrams were created using the ‘UpSetR’ (V. 1.4.0) and venneuler packages (X.x). Area-proportional Venn diagrams of genus lists, with 20 reads per million total reads and eukaryotic genera-only cutoffs, were generated using BioVenn70.
Human exome-based variant analysis
The Illumina human exome variant analysis was performed using the nf-core Sarek pipeline71, leveraging Nextflow for workflow management and Docker for consistent execution environments. The preparation for base quality score recalibration involved generating the necessary files required for the recalibration process using the GATK.GRCh38 reference genome. This step ensured that the recalibration could be accurately performed, correcting any systematic biases introduced during sequencing. The base quality scores of the sequencing reads were recalibrated to adjust for any biases introduced during the sequencing process. This recalibration, performed using the GATK.GRCh38 reference genome, improved the accuracy of the quality scores, leading to more reliable variant calling. Variant calling was performed using the HaplotypeCaller tool from GATK, with the GATK.GRCh38 reference genome. This step involved identifying potential variants, including SNPs and insertions/deletions (indels), from the recalibrated sequencing data. The coordinates from the phase 1 high confidence SNPs, and known indels at https://genome.ucsc.edu/cgi-bin/hgTrackUi?db=hg19&g=tgpPhase1 were used to filter the variant calls. The identified variants were then outputted in VCF format for further analysis. The final step involved annotating the identified variants using the Variant Effect Predictor with the GATK.GRCh38 reference genome. This annotation process provided functional information about the variants, such as their potential impact on genes and proteins, which aids in the interpretation of their biological importance.
Assessment of reference genome specificity and read clustering of ONT eDNA reads
Nanopore eDNA reads were prefiltered via the CZ ID ONT mNGS pipeline v0.7. We then used minimap v2.26 (ref. 65) to align 6.5 million reads (Forest air April 2023) and 1.4 million reads (Mountain air July 2023) (‘sample.humanfiltered.fastq’) against a set of 45 publically available reference genomes from NCBI RefSeq (Supplementary Table 2). To provide a more stringent categorization and further minimize human sequence contamination risks, we also aligned reads against the human telomere-to-telomere (T2T-CHM13v2.0) assembly63. All resulting files were indexed, binarized and sorted using samtools v1.17 (ref. 72). We extracted the IDs of all nanopore sequences mapping to any of the 45 species but not T2T-CHM13v2.0 (378,856 reads, 5.8% of the original set) by use of samtools v1.17, before generating a binary presence/absence matrix of reads versus reference genomes in R. Given the large size of this matrix, we undertook two hierarchical clustering experiments on down-sampled representations of nanopore reads: (1) group pruning: for a pruned representation of the sequences mapping to different individual species or shared subsets of reference genomes, we reduced each fraction to a maximum of ten reads, thereby reducing the dimension to a total of 1,100 reads, and (2) random sampling: to provide a quantitative perspective of the different set sizes, we uniformly sampled 1,100 reads from the original matrix. The matrix entries were then hierarchically clustered and displayed using the pheatmap function in R.
Metabarcoding analysis
The metabarcoding 12S rRNA gene sequence reads were initially examined for quality scores with FastQC61 and primers were trimmed using Cutadapt73. All sequences were subsequently processed using the DADA2 pipeline74. Briefly, reads were filtered out (maxN = 0, maxEE = c(2, 2), truncQ = 2, minLen = 50), and remaining unpaired sequences were then denoised and aligned. The resulting ASV table was then cross-referenced against the 12S Vertebrate North American Reference Set75 (https://doi.org/10.5281/zenodo.7888133), using the ‘blast_nucleotide_to_nucleotide’ function in the ‘metablastr’ R-package76, with a >99% identity cut off.
qPCRFlorida
A pan-eukaryotic 18S ribosomal RNA (rRNA) gene (Applied Biosystem, 4352930E) prevalidated Taqman Gene Expression assay, which also has both primers and probe within a single exon (that is, can be used to detect genomic DNA or complementary DNA), was used to quantify the total level of pan-eukaryotic gDNA in samples6,7,8,53. The qPCR reaction mixtures were performed on 384-well plates in a total volume of 10 μl per well: 5 μl TaqMan Fast Advanced Master Mix (Fisher Scientific, cat. no. 4444557); 3.5 μl Nuclease-free water (Fisher Scientific); 0.5 μl of the 18 s rRNA assay (at manufacturer-supplied concentration); 1 μl DNA template (or, for no-template controls, an additional 1 μl of nuclease-free water per well). Each air sample or NFC was run in three technical replicates. No-template controls were run in triplicate on every qPCR plate. The qPCR reactions were performed on an Applied Biosystems QuantStudio 6 Pro with the following cycling parameter: 95 °C for 20 s for one cycle, followed by 30 cycles of 95 °C for 1 s and 60°C for 20 s. Serial dilution of total DNA that had previously been extracted from green sea turtle (Chelonia mydas) blood was used as the template for the standard curve. The serial dilution range was 621 ng μl−1 to 0.00621 ng μl−1, with a 1-μl template used per well.
Ireland
The Applied Biosystem prevalidated Taqman Gene Expression qPCR assay directed against the human ZNF285 gene (assay ID Hs00603276_s1) was used as a species-specific human assay, based on having no cross-reactivity with over 29 other species from mice to plants (https://www.thermofisher.com/order/genome-database/ and refs. 7,31) and having both primers and probe within a single exon (that is, detect DNA). A pan-eukaryotic 18S rRNA gene (Applied Biosystem, 4319413E) prevalidated Taqman Gene Expression assay, which also has both primers and probe within a single exon (that is, detects DNA), was used to quantify the total level of pan-eukaryotic DNA in each of the Irish samples7. The qPCR reaction mixtures were performed on 384-well plates in a total volume of 10 μl per well: 5 μl TaqMan Gene Expression Master Mix (Fisher Scientific, cat. no. 4369016), 3.5 μl nuclease-free water (Fisher Scientific), 0.5 μl of the respective assay (primer/probe mix, manufacturer-supplied concentration) and 1 μl DNA template (or, for no-template controls, an additional 1 μl of nuclease-free water per well). Each biological sample, NFC and standard curve point was run in three technical replicates. No-template controls were run in triplicate on every qPCR plate. The qPCR reactions were performed on an Applied Biosystems QuantStudio 7 Flex, at the Conway Institute’s Genomics Core, University College Dublin, with the following cycling parameter: 50 °C for 2 min and 95 °C for 10 min for 1 cycle, followed by 45 cycles of 95 °C for 15 s and 60 °C for 1 min. For absolute quantity qPCRs, a standard curve was generated using six DNA one-in-ten serial dilutions, ranging from 111.3 ng μl−1 to 0.001113 ng μl−1. A 1-μl template was used per standard curve reaction. The standard curve template used for both pan-eukaryotic and human-specific qPCRs was DNA extracted from cultured human IMR32 neuroblastoma cells in May 2013 for the Duffy et al. 2018 study77. For each standard curve concentration and each eDNA sample, technical triplicate wells/reactions were performed.
qPCR results were plotted with BoxPlotR78 (http://shiny.chemgrid.org/boxplotr/) with every datapoint displayed. Tukey whiskers (extend to data points that are less than 1.5× interquartile range away from the first and third quartile) were utilized for every boxplot. One box is graphed per single sample, consisting of all qPCR technical replicate wells for that sample.
Box 1 The Promethean dilemma of recovering human genetic material from the air: considerations of using human eDNA for the public good or facilitating surveillance states
We obtained ethical approvals for this current air eDNA study. Although we first highlighted the issue in 2023 and called for stakeholder, regulator and public action7, no regulations have yet been established regarding the collection of human genetic information from environmental samples. Here, we highlight that such considerations must now also extend to outdoor airborne DNA, particularly in areas of high human habitation. As with our previous study, we intentionally refrained from attempting individual human identification, despite having long-read data, since our initial call to action7 for regulations to be established was relatively recent.
The ease with which human genetic information is recoverable from even outdoor air eDNA further highlights the important ethical considerations we recently raised7 and which are beginning to be considered7,79,80,81. Given the ready feasibility reported here of human genomic analysis from air eDNA shotgun sequencing (whole-genome sequencing) and human whole-exome enrichment and sequencing, all the ethical and privacy implications previously considered for water, sediment and indoor air7,79,80,81 must be extended to outdoor air eDNA as a matter of urgency. The current findings can be used to inform policy discussion, while also highlighting the applicability of eDNA genomics for both humans and non-humans.
Attempts to unanimously agree on international policies on generative artificial intelligence use have recently failed82. Similarly, a call for a voluntarily moratorium80 on human eDNA investigations has yet to garner widespread support. A blanket embargo ignores the possibility of bad faith actors/unregulated countries and ignores the beneficial medical, environmental and law enforcement applications7,36. For example, forensic science continues apace to utilize human DNA recovered from environmental substrates (eDNA); indeed, individual-level identification has already been achieved83,84,85,86,87,88. Despite using similar approaches, wildlife eDNA researchers are unlikely to be qualified or mandated to dictate forensics policies (as would be a consequence of a blanket embargo). It may be less consequential for non-human-focused fields, where human eDNA is only bycatch7, to implement a moratorium. Targeted policies are required, such as regulation of who should directly investigate human eDNA, when and where such investigation is conducted and what approvals are needed. Other embargo alternatives include molecular-based blocking of human DNA before sequencing for non-human-focused studies and/or automatically filtering human-aligning reads out of datasets before they are visible to investigators or submitted to public repositories7,79,81. Note that no control/management approach is without its own complications, and each has benefits and drawbacks7. In addition, the thorny issue of conflicting policies in different jurisdictions (or, indeed, international waters) and the fact that eDNA shed in one jurisdiction can be transported in the air or flowing water to other jurisdictions89 need to be considered.
High-quality airborne DNA recovery, as reported here, coupled with existing single-cell/single-nucleus sequencing technologies, provide seriously powerful potential for individual-level surveillance of flora, fauna, funga and also humans, so should be proactively considered by regulators. As with artificial intelligence technologies, the human eDNA genie cannot be returned to the bottle. Therefore, consideration by regulators, public and stakeholders, responsible informed policy discussions, and implementation with broad spectrum participation are required.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.