Papillomaviruses have been identified in a wide array of vertebrates. With over 240 distinct types classified in 37 genera, papillomaviruses are one of the most successful families of vertebrate viruses [30]. They have been, and continue to be, an astonishing evolutionary success. HPV 33 and 35 are among the least phylogenetically studied despite their classification as high-risk types and group 1 carcinogen status [30, 31]. Both genotypes belong to the family of Alpha-papillomaviridae, specifically the Alpha 9 species. The International Agency for Research on Cancer (IARC) concluded that these genotypes have consistent and sufficient epidemiological, experimental and mechanistic evidence of carcinogenicity in humans for cervical cancer, as well as the HPV 31, HPV 39 and HPV 45 [31, 32]. Other carcinogenic HPV types are also implicated in cervical cancer, although they are less frequently detected compared to HPV 16, 18, 31, 39, and 45 [32].
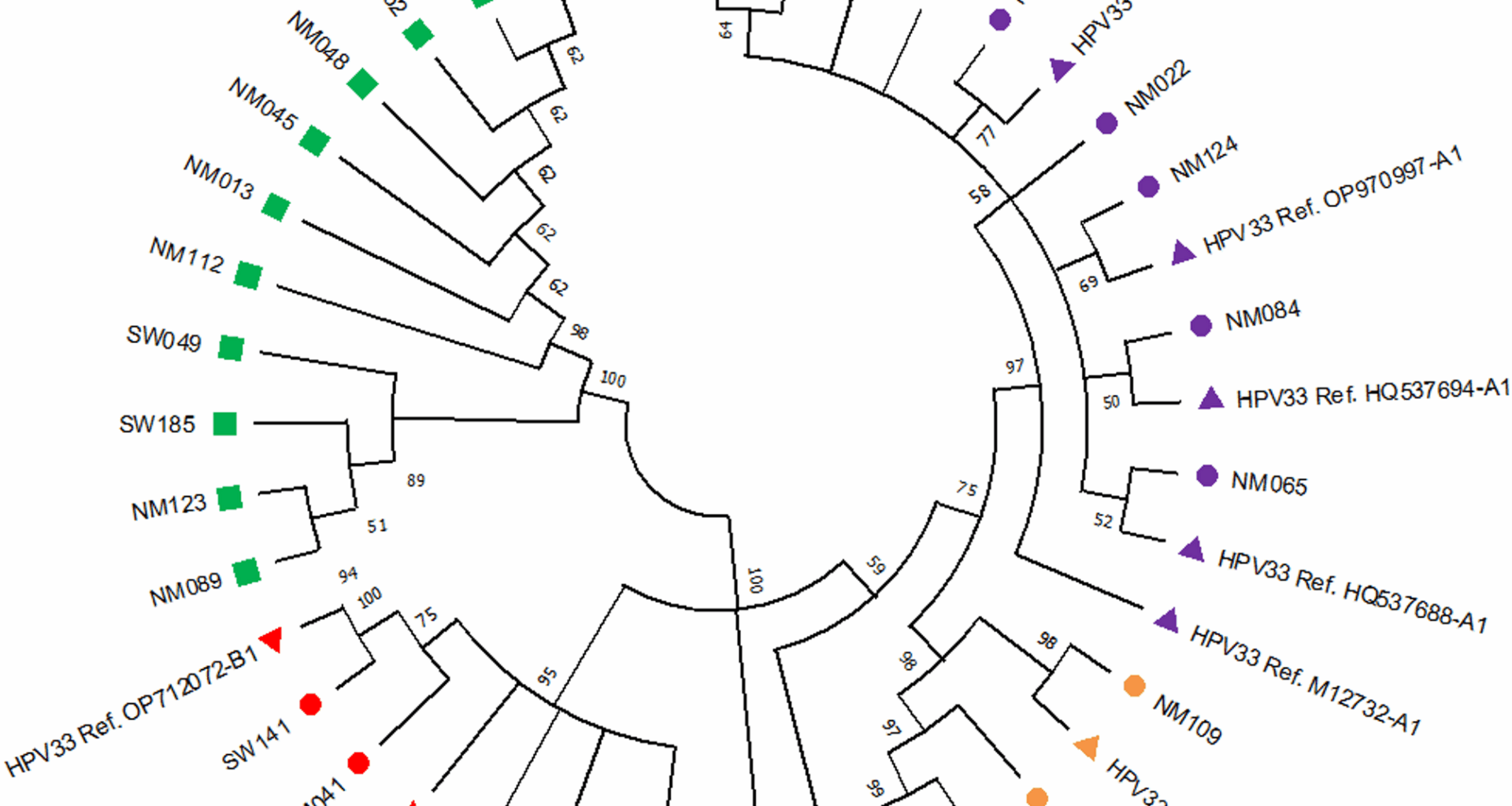
In this study, 62% of HPV 33-positive participants were women with HSIL, confirming previous reports linking HPV 33 to severe cervical abnormalities. In sub-Saharan Africa, there is a lack of studies conducted to characterise the HPV 33 relationship with cervical abnormalities. Some studies conducted in Asia showed that in some cases, HPV 33 is more prevalent than HPV 18 in women with cervical lesions, being the second most common after HPV 16 [33, 34]. In the study conducted by Wang et al. (2010) on 1387 women diagnosed with cervical intraepithelial neoplasias (CIN 2 +), where hr-HPV positivity was found to be 91.6%, the types most commonly associated with CIN 2 + was listed as HPV 16, 58, 33 with 59.3%, 14.4%, and 10.0%, respectively [35]. In a study conducted by Purut and Uçkan (2023), on women with cervical intraepithelial lesions, the colposcopy results showed HPV 16 positivity in 43.3%, HPV 33 positivity in 30% and HPV 18 positivity in 10% of the patients with CIN2 + and above lesions [33].
Additionally, HPV 33 positivity has been linked to a higher incidence of CIN2 + lesions compared to lower-grade abnormalities [41]. Some studies indicate that HPV 33 is an independent predictor of CIN2 + lesions, with a fivefold increase in CIN2 + risk among HPV 33-positive women [41, 42]. This supports the hypothesis that HPV 33 plays a significant role in cervical malignancy development [42, 43]. In this study, HPV 33 European variants (A1 and A2 sublineages) were the most prevalent (38%), with the A1 sublineage accounting for 31%. The African 2 variant (B1 sublineage) was found in 19% of cases. Previous studies have identified A and B as the two predominant HPV 33 lineages [7, 11]. A study by Chen et al. (2013), which analysed HPV 33-positive cervical samples from the International Agency for Research on Cancer (IARC) biobank, found that A1, A2 and B sublineages were the most prevalent [11]. The A1 sublineage was strongly over-represented in cervical cases compared to controls in both Africa and Europe [11]. In 2011, Chen et al. sequenced the complete 8 kb genomes of 120 alpha-9 types (HPV 31, 33, 35, 52, 58 and 67) in samples collected from women from different African, Asian and American countries, to capture maximum viral heterogeneity and variations [10]. The results obtained for HPV 33 positive cases were the same as our present study results, where most of the study subjects clustered into the A1 sublineage.
In this study, the HPV 33 E6 region had twice as many SNPs as observed in the E7 region, and those SNPs were mostly found in the 11 samples that did not cluster with any of the reference sequences under investigation. Previous studies suggested that the HPV 33 E7 region is more conserved than E6 and, as a result, is considered a more suitable target for the diagnostic detection of HPV 33 [7, 19]. The intratypic variations observed in HPV 33 E6 and E7 can provide helpful information for identifying and distinguishing the known and the new HPV genotypes.
HPV 35 is frequently identified in women with cervical dysplasia and cervical cancer across multiple regions, including America, Asia and sub-Saharan Africa [12, 13, 36]. Consistent with previous research, our results confirm that HPV 35 exhibits lower genetic variation compared to other hrHPV types [31]. Consistent with the low level of genetic variation, HPV 35 is only divided into two sublineages, A1 and A2, rather than separate variant lineages, which are defined as requiring greater than 1.0% difference between complete genomes [37].
In this study, 78% of HPV 35 isolates clustered within the A1 sublineage, with 38% of these found in women with HSIL. Several studies have evaluated the association of HPV 35 A1 sublineage with the risk of cervical abnormalities. For example, a 7-year longitudinal study in Costa Rica found that the HPV 35 A1 sublineage may be associated with a higher risk of persistence and the development of CIN3 + compared to the A2 sublineage [35].
However, a study in the U.S. following up women every six months for two years identified no difference in cervical lesion risk between HPV35 sublineages [38]. In a Zimbabwean HPV 35-positive cohort, two of the five whole-genome sequences associated with HSIL clustered within the A2 sublineage [38]. Thus, it was suggested that future large-scale studies including HPV whole genome sequencing would be required to determine whether differential cervical cancer risk is associated with HPV 35 sublineages, and/or individual viral variants, including single-nucleotide changes and insertions/deletions [38, 39, 40].
It is suggested that the relationship between the European A2 lineages with the women of African ancestry could be a consequence of thousands of years of virus-host interactions, where certain lineages of HPV 35 became better adapted and able to persist in these women. Additionally, a likely explanation is that a genetic bottleneck due to the different out-of-Africa events and/or gene introgression events from archaic human populations may have rendered modern humans of non-African ancestry less susceptible to infection and/or progression with HPV35, and specifically the A2 sublineage [31].
Contrary to the high genetic variation observed between variants of other high-risk HPV types, the low phylogenetic distance between HPV 35 variants presented in this study is consistent with what is commonly observed in other populations around the world, with most HPV 35 variants clustering between only two sublineages (A1 or A2 sublineage) [10, 32]. Furthermore, in our study subjects were observed a significant diversity in the genealogical distribution since they are clustered among the A1 sublineages of the HPV 35 reference sequences isolated in countries such as Zambia, South Africa, Costa Rica, Togo and Zimbabwe.
HPV 35 is one of the most genetically conserved hrHPV types since its genome is relatively stable compared to other hr-HPV genotypes [10, 39]. In this study, three E6 SNPs (127 T > C, 136 T > C, and 341 T > C) were the most prevalent (39%) and were previously identified in other studies conducted in populations from the Americas, Asia, and Africa [31, 38, 40, 41]. In the E7 region, only one SNP (675 T > C) was found commonly. However, none of these SNPs were previously described or known to affect oncogenicity or immune escape.
This study found significant genetic variation in the HPV 35 E6 oncogene compared to the E7, a finding previously observed in several studies. For example, Mboumba Bouassa et al. (2024) explored the genetic variability of clinical strains of HPV 35 isolated from HIV-negative heterosexual adult women living in N’Djamena, Chad and HIV-infected men having sex with men (MSM) in the Central African Republic, where HPV 35 is highly prevalent. Their findings demonstrated that HPV 35 exhibited a higher rate of genetic variability in the E6 oncogene, while E7 sequences were highly conserved [40].
In this study, the most prevalent SNPs in the E6 gene were primarily found in isolates that did not cluster with any of the investigated reference sequences. Most of these SNPs were previously detected in A2 HPV 35 sublineages in whole-genome sequencing studies [38, 39, 40]. Evolutionary divergence in HPV variant lineages is common and is associated with different cancer risks, ethnicity, and geographical distribution [10, 30]. However, it is well established that the HPV 33 and 35 genotypes share a common ancestor with HPV 16, making them related within the same genotypes’ clade [10].
HPV 16 and HPV 18 sublineages have also been associated with cervical precancer/cancer in certain ethnic groups [42], and a viral-host interaction has been posited for HPV 16, where both modern-human migration and reproductive events, with introgression of immune-related alleles, confer a host niche adaptation, potentially influencing phenotype, such as cervical cancer [42, 43]. To add to this, Phylogenetic analysis in African HPV sequences demonstrated that the African HPVs share genetic ancestry with European sequences, whereas American isolates are less closely related. Migration analysis revealed a significant asymmetry in HPV flow, with migration rates from Africa to Europe [44].
Some study isolates did not cluster with any of the reference sequences under investigation, highlighting the need for whole-genome sequencing to improve variant characterisation [45]. Our findings were strictly based on a portion of the HPV genome. This fact does not allow us to classify the non-clustering subject as a novel HPV sublineage, recombinants, as well as to define whether the SNPs could be an immune escape, or they can interfere and impact the current HPV vaccines. However, due to the importance of E6 and E7 as oncogenes, future studies are needed to clarify the potential function of these variants. Additionally, our relatively small sample size limits statistical power, and future studies with larger cohorts could provide stronger associations between HPV 33 and HPV 35 sublineages and cervical cancer risk. This is a cross-sectional study, and this fact could be a limitation for the causal inference between variants and lesion severity in the studied subjects. Furthermore, our study subjects were collected only in two countries (South Africa and Mozambique), a fact that represents a geographical scope limitation, hence reducing our findings.
The low genetic diversity of HPV 35 isolates aligns with previous studies. However, given that some isolates in the present study did not cluster with the reference sequences, we speculate that whole-genome sequencing analysis would give a comprehensive understanding of this genetic diversity.
This study confirms a high prevalence of European HPV 33 and 35 variants. Taking into consideration that HPV 35 is still not targeted by the current HPV vaccines, further research is required to consider whether the addition of HPV 35 to the next generation of vaccines would result in a meaningfully broader coverage for African women. Vaccination and screening approaches that cover all the highest-risk types are needed for optimal global prevention coverage. Taking into consideration the present study’s genetic diversity evidence associated with a high burden of cervical cancer in Africa, prevention programs in populations without quality screening, the addition of HPV 35 to HPV vaccines would result in improved cervical cancer prevention. The current study findings add to the information on the variation in E6 and E7 of two important HPV types known to play an essential role in a significant number of African cervical cancers. This will help to inform the design of therapeutic vaccines for this region.