More than 4,000 genes have been established as etiological for a rare disease, of which only 69 are noncoding1. Three of these noncoding genes—RNU4ATAC, RNU12 and RNU4-2—encode snRNAs that have crucial roles in pre-messenger RNA (mRNA) splicing. Variants in RNU4ATAC are responsible for microcephalic osteodysplastic primordial dwarfism type I (refs. 2,3), Roifman syndrome4 and Lowry–Wood syndrome5, whereas variants in RNU12 cause early-onset cerebellar ataxia6 and CDAGS syndrome7. These pathologies are inherited in an autosomal-recessive manner. Both RNU4ATAC and RNU12 encode components of the minor spliceosome, a molecular complex that catalyzes splicing for fewer than 1% of all introns in humans8. However, more than 99% of introns are spliced by the major spliceosome. Recently, we reported that de novo mutations in RNU4-2, which is transcribed into the U4-2 snRNA component of the major spliceosome, cause one of the most prevalent monogenic neurodevelopmental disorders (NDDs)9. The discovery was published independently by a separate group10.
To explore whether other noncoding genes might also be causal for NDDs, we performed a refined statistical analysis of the 100,000 Genomes Project (100KGP) data in the National Genomic Research Library (NGRL)11. Following a previously described approach9,12, we used the BeviMed genetic association method13 to compare rare variant genotypes in the 41,132 canonical transcript entries in Ensembl v.104 with a biotype other than ‘protein_coding’ (Supplementary Data), which included 14,307 entries annotated as pseudogene transcripts, between 7,452 unrelated, unexplained cases annotated with the ‘Neurodevelopmental abnormality’ (NDA) Human Phenotype Ontology (HPO) term and 43,727 unrelated participants without the NDA term. Notably, whereas our previous analyses filtered out single-nucleotide variants with combined annotation-dependent depletion (CADD)14 score
Our analysis yielded only two genes with a posterior probability of association (PPA) with NDA > 0.5. RNU4-2, which we have reported previously9, had a PPA of ~1, and RNU2-2P (now called RNU2-2) had a PPA of 0.97. The association with RNU2-2 depended on inclusion of variants with CADD scores ≤ 10 (Extended Data Fig. 1). Conditional on the association, two variants, at nucleotide positions 4 and 35, had a BeviMed posterior probability of pathogenicity (PPP) > 0.5 (Fig. 1a). The nine NDA cases with either of the variants had a significantly greater phenotypic homogeneity based on HPO terms than expected under random selection of nine NDA cases from unexplained and unrelated NDA study participants in the 100KGP (P = 1.33 × 10−3, Fig. 1b), supporting causality for a distinct NDD. RNU2-2 has a 191-bp sequence that is identical to that of the canonical gene RNU2-1, except for eight single-nucleotide substitutions (all within n.108–191). Unlike RNU2-1, which has a variable copy number within a region on chromosome 17, RNU2-2 has a unique sequence occurring in only one location on chromosome 11. Although at the time of analysis, RNU2-2 was known as RNU2-2P and annotated as one of many U2 pseudogenes in bioinformatics databases15, it has recently been shown to be expressed in cell lines, and its transcripts, U2-2P (now U2-2), have been shown to have the greatest abundance and stability of all noncanonical U2 snRNAs16. After aggregation over the 11 copies of RNU2-1 in the GRCh38 build of the reference genome, RNU2-1 and RNU2-2 show comparable levels of expression in whole blood and in blood cells (Fig. 1c). RNU2-2 resides in a 5′ untranslated exon of WDR74 that had previously been identified as being enriched for hotspot mutations in cancer, although the existence of RNU2-2 at that locus was not known at the time17. A recent study showed that both RNU2-1 and RNU2-2 carry recurrent somatic mutations (n.28C>T) that drive B cell-derived tumors, prostate cancers and pancreatic cancers18. The same study showed that RNU2-2 is a functional gene that is transcribed independently of WDR74—a finding that we recapitulated in blood and blood cells (Extended Data Fig. 2)—and that both the canonical U2-1 and noncanonical U2-2 snRNAs are incorporated into the spliceosome18.
Fig. 1: Discovery and replication of RNU2-2 as an etiological gene for a new NDD.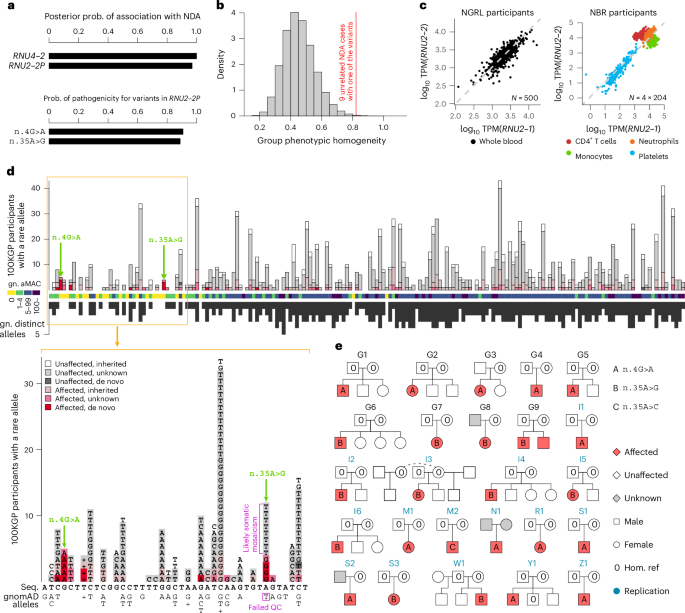
a, BeviMed PPAs between each of RNU4-2 and RNU2-2 (previously known as RNU2-2P) and NDA. All other noncoding genes and pseudogenes had PPA RNU2-2 variants had conditional PPP > 0.5: n.4G>A and n.35A>G. Prob., probability. b, Distribution of phenotypic homogeneity scores for 100,000 randomly selected sets of nine participants chosen from 9,112 unrelated NDA-coded participants. The score corresponding to the nine identified cases with one of the two RNU2-2 variants with PPP > 0.5 is indicated with a red line. c, Scatter plot of log10 expression of RNU2-1 against that of RNU2-2 in whole-blood samples from a random subset of 500 participants in the NGRL and in four blood cell types from 204 NBR participants. TPM, transcripts per million. d, Top, numbers of participants with a rare allele at each of the 191 bases of RNU2-2, stratified by affection status and inheritance information of the carried allele. The two variants with PPP > 0.5 are indicated with green arrows. The color-coded track shows the aggregated (over distinct alleles at a position) minor allele count (aMAC) in gnomAD v.4.1.0 (gn.) at each position, and the black bars show the numbers of distinct alternate alleles in gnomAD at each position (multiple insertions and multiple deletions at a given position each count as one). Variants failing quality control (QC) in gnomAD are not shown in this subpanel. Bottom, data corresponding to nucleotide positions 1 to 41 in greater detail, including gnomAD-QC-failing variant n.35A>T. Above and below the RNU2-2 cDNA sequence (Seq.), the alternate alleles in 100KGP participants and the distinct alleles in gnomAD are shown, respectively; ‘+’ indicates insertions, and the variant that failed QC in gnomAD is indicated. e, Pedigrees for participants with a rare alternate allele n.4 or n.35 in RNU2-2. Pedigrees used for discovery have a ‘G’ prefix and are labeled in black. Pedigrees used for replication in the IMPaCT-GENóMICA, URDCat and ENoD-CIBERER aggregate collection; the 100KGP; the NBR; Erasmus MC UMC; the GMS; Radboud UMC; deCODE or the ZOEMBA study have an ‘I’, ‘M’, ‘N’, ‘R’, ‘S’, ‘W’, ‘Y’ or ‘Z’ prefix, respectively, and are labeled in blue. Hom., homozygous; ref., reference.
The two germline variants with a high PPP, n.4G>A and n.35A>G, are located in a genomic locus spanning a region of approximately 40 nucleotides at the 5′ end of the 191-bp RNU2-2 gene. The locus has a markedly reduced density of population genetic variation in gnomAD19, consistent with the effects of negative selection (Fig. 1d). Published secondary structure data of the U2 snRNA show that r.4 is located within the helix II U2–U6 interaction domain, whereas r.35 is part of the highly conserved recognition domain GUAGUA that binds the branch sites of introns20,21,22 (Extended Data Fig. 3). Trio sequencing of four of the five cases with n.4G>A and three of the four cases with n.35A>G showed that the variants were de novo in each case. A variant with a different alternate allele at nucleotide 35, n.35A>T, was called in eight unaffected participants; it was also present in gnomAD but failed quality control (QC) (Fig. 1d). Analysis of whole-genome sequencing (WGS) and Sanger sequencing data suggested that n.35A>G is a germline variant, but n.35A>T is a recurring somatic mosaic variant. This somatic variant is observed only in individuals over the age of 40 years, consistent with clonal hematopoiesis (Extended Data Fig. 4).
To replicate our findings in the nine NDD cases, we examined eight additional rare disease collections: a component of the 100KGP not included in the discovery dataset (10,373 participants, of whom 1,736 have an NDA); the NIHR BioResource-Rare Diseases (NBR) data23 (7,388 participants, of whom 731 have an NDA); the UK Genomic Medicine Service (GMS) data (32,030 participants, of whom 6,469 have an NDA); data from the Erasmus MC UMC (1,527 participants, of whom approximately 400 have an NDA); an aggregate of the IMPaCT-GENóMICA, URDCat and ENoD-CIBERER programs for undiagnosed rare diseases24 (1,707 probands with NDDs and WGS data); clinical data from Radboud UMC Nijmegen (1,037 probands with an NDA); WGS data from deCODE genetics (73,821 participants, of whom 4,416 have an NDA) and data from the ZOEMBA study (127 participants, of whom 71 have an NDA). We identified a further 16 cases in these replication collections (Fig. 1e), all but two of whom were confirmed to have a de novo variant. There were no unaffected carriers of either variant. Eight replication cases had n.4G>A, seven replication cases had n.35A>G, and one replication case had a different alternate allele at nucleotide 35, n.35A>C. Although this case represented the only individual harboring n.35A>C, modeling of the interactions between U2-2 snRNA and canonical branch site sequences suggested that n.35A>C has a destabilizing effect on binding that is greater than that of the n.35A>G variant and in many cases similar in magnitude to that of the n.4G>A variant with respect to its cognate partner U6 (Extended Data Fig. 5). All these variants were called confidently by WGS (Extended Data Fig. 6). In the 100KGP, RNU2-2 was a more prevalent etiological gene than all but 29 of the ~1,400 known etiological genes for intellectual disability, explaining about one-fifth the number of cases as RNU4-2, the etiological gene for RNU4-2 syndrome, also known as ReNU syndrome (Fig. 2). This relative prevalence was consistent with observations in the IMPaCT-GENóMICA, URDCat and ENoD-CIBERER aggregate collection, which identified 27 cases with RNU4-2 syndrome and six cases (that is, 4.5 times fewer) with RNU2-2 syndrome.
Fig. 2: Prevalence in the 100KGP.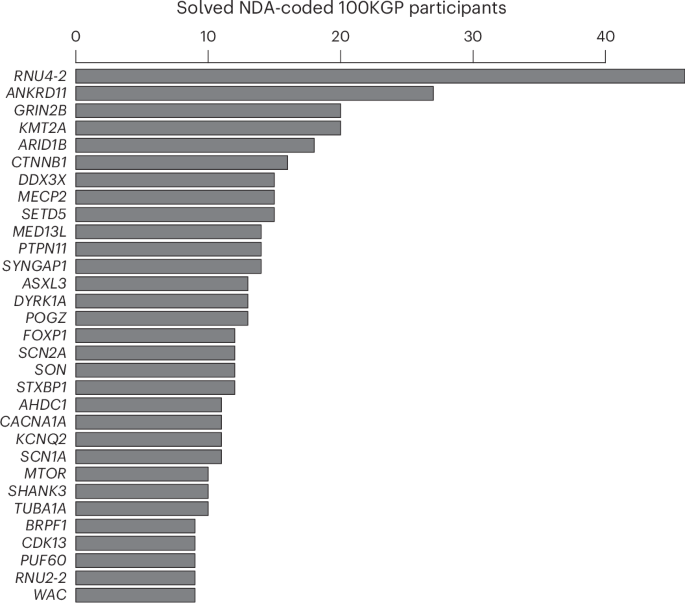
Of the 9,112 unrelated NDA-coded cases in the 100KGP, the numbers solved through pathogenic or likely pathogenic variants in a gene are shown, provided at least nine cases were diagnosed. For RNU2-2, the number of NDA-coded cases in the 100KGP with one of the recurring de novo variants is shown.
Analysis of HPO terms for the nine uniformly phenotyped 100KGP cases revealed that 100% were assigned ‘Intellectual disability’ and ‘Global developmental delay’, 89% were assigned ‘Delayed speech and language development’, 78% were assigned ‘Motor delay’ and 56% were assigned ‘Autistic behavior’, in line with frequencies among NDA cases generally (Fig. 3). However, certain terms were enriched in RNU2-2 cases: ‘Seizure’ was annotated in 89% of RNU2-2 cases (versus 27% in other NDA cases, Bonferroni-adjusted (BA) P = 2.44 × 10−3) but later confirmed to be present in 100%, ‘Microcephaly’ in 78% of cases (versus 18%, BA P = 1.62 × 10−3), ‘Generalized hypotonia’ in 56% of cases (versus 13%, BA P = 3.56 × 10−2), ‘Severe global developmental delay’ in 44% (versus 2.7%, BA P = 8.89 × 10−4) and ‘Hyperventilation’ in 33% of cases (versus 0.16%, BA P = 7.56 × 10−6). No HPO terms were significantly underrepresented in the RNU2-2 cases. Of the terms that were enriched among cases of RNU4-2 syndrome, ‘Seizure’, ‘Microcephaly’ and ‘Generalized hypotonia’ were also enriched in RNU2-2 cases. However, ‘Severe global developmental delay’ and ‘Hyperventilation’ were only enriched in RNU2-2 cases, suggesting that these may be differentiating phenotypic features. Strikingly, three RNU2-2 cases were coded with the seldom-used ‘Hyperventilation’ term by three independent clinicians.
Fig. 3: Phenotypic enrichment in the 100KGP.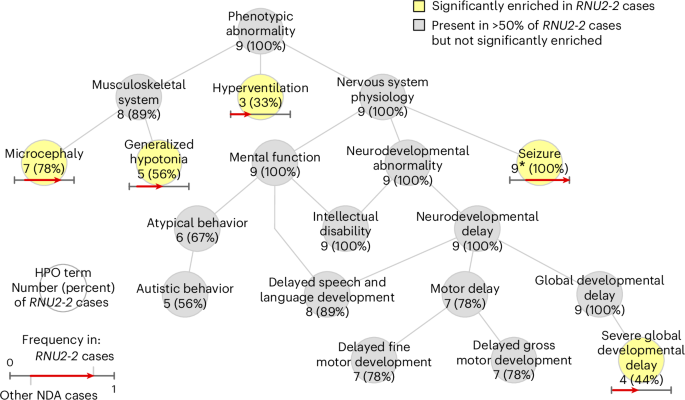
Graph showing the ‘is-a’ relationships among HPO terms present in at least three of the nine NDA-coded RNU2-2 cases in the discovery collection or significantly enriched among them relative to 9,112 unrelated NDA-coded participants of the 100KGP. The significantly overrepresented terms are highlighted. For each term, the number of cases with the term and the proportion that number represents out of nine is shown. For each overrepresented term, the proportion of NDA-coded participants with the term and the proportion of NDA-coded RNU2-2 cases with the term are represented as the horizontal coordinate of the base and the head of an arrow, respectively. *, Only eight of the nine (89%) of the cases had the ‘Seizure’ HPO term in the NGRL, but epilepsy was confirmed in the case without the HPO term by inspecting the individual’s electronic health record and the numbers attached to ‘Seizure’ were updated accordingly.
Detailed clinical vignettes for the 15 cases in pedigrees G1–2, G4, I1–6, M2, R1, S3, W1, Y1 and Z1 are provided in Supplementary Note and Supplementary Table 1. These indicate that the neurodevelopmental phenotype caused by the RNU2-2 variants typically manifests from 3 to 6 months of age but is progressive, frequently severe and accompanied by characteristic dysmorphic features (Fig. 4). All the cases displayed prominent epilepsy, usually from the first few months of life, and seizures were severe and pharmacoresistant. Seizures were characteristically complex and included spasms, tonic, tonic clonic, myoclonic and absence types, classified in some probands as Lennox–Gastaut syndrome. These features distinguish the RNU2-2 cases from previously reported cases of RNU4-2 syndrome, in which the developmental phenotype was reported as less severe, some of the dysmorphic features were different, and epilepsy was typically later in onset, less severe and more commonly focal9,10,25. Extraordinarily, case M2 also harbored a de novo truncating variant in SPEN predicted to cause Radio–Tartaglia syndrome26. However, the individual in this case had short stature (RNU2-2 patients than Radio–Tartaglia syndrome patients (Supplementary Note). This atypical presentation is consistent with a dual rare genetic diagnosis.
Fig. 4: Clinical photographs.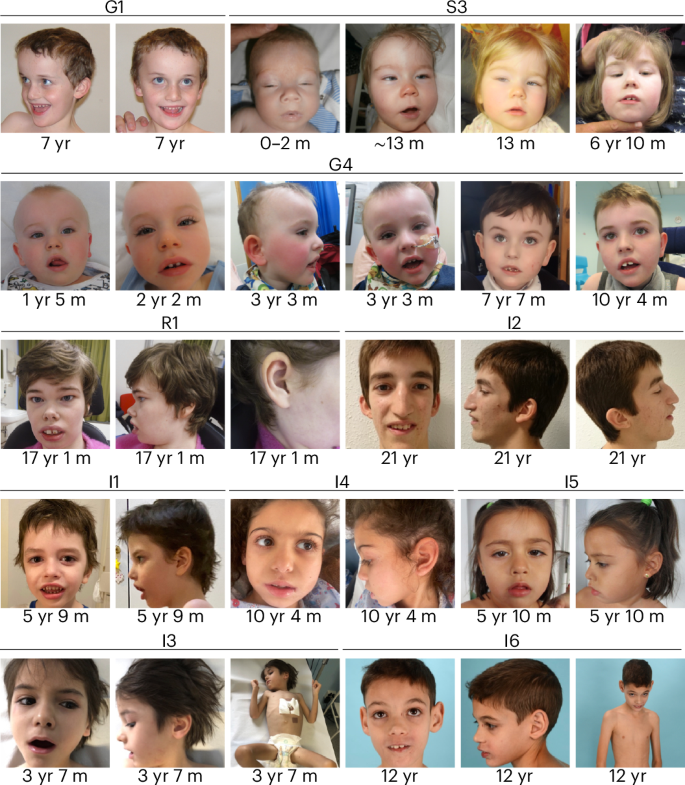
Clinical photographs of individuals from pedigrees G1, G4, S3, R1 and I1–6. The individuals in these cases show common features of long palpebral fissures with slight eversion of the lateral lower lids, long eyelashes, broad nasal root, large low set ears, wide mouth and wide spaced teeth. The approximate ages of the individuals when the photographs were taken are shown. Photographs of individual M2, who has Radio–Tartaglia syndrome in addition to RNU2-2 syndrome, are included in the Supplementary Note. We have obtained specific consent from the families to publish these clinical photographs. m, months; yr, years.
Using trio WGS data, which were available for 17 families, we were able to determine the parental origin of the de novo mutations for ten of those families. Echoing observations in cases with RNU4-2 syndrome, the pathogenic RNU2-2 mutations were ubiquitously of maternal origin, suggesting that they may affect spermatogenesis. Analysis of uniquely aligned reads at heterozygous sites in whole-blood RNA sequencing (RNA-seq) data revealed that both alleles of RNU2-2 were expressed robustly in cases (Extended Data Fig. 7). However, a genome-wide comparison of the RNA-seq alignments between five cases and 495 unrelated unexplained NDA-coded participants did not reveal differential gene expression, differential splice junction usage or any pattern of aberrant splicing in the cases (Extended Data Fig. 8), suggesting that transcriptomic analysis of other tissue types will be required to uncover the underlying molecular mediators of disease.
U2 is involved in all stages of pre-mRNA splicing and contains distinct domains that interact with the catalytic U6, intronic branch sites and scaffolding of several protein assemblies27. Notably, the U6 binding domain and the branch site recognition domain of U2-2 are transcribed from a region in RNU2-2 exhibiting markedly reduced population genetic variation (Fig. 1d). Studies in the 1990s of yeast U2 snRNA showed that variants in branch site recognition sequence GUAGUA inhibit splicing and even generate a dominant lethal phenotype when the recognition sequence is changed entirely28,29. Position r.35 in the human U2 sequence corresponds to r.36 in the yeast U2 sequence, where n.36A>G and n.36A>T result in 0–10% and 10–20% splicing activity, respectively, compared with the wild-type sequence29. Although the U2–U6 recognition sequences are not conserved between yeast and human, a similar organization is retained. The U2–U6 interaction in yeast is not very sensitive to variation in U2 snRNA29, but genetic suppression experiments that changed multiple residues within U2 or U6 snRNAs, including position r.4 in U2 snRNA, have demonstrated that the U2–U6 helix II plays a part in the regulation of splicing in mammalian cells30,31. Mice with variants in a direct ortholog of RNU2-2 do not exist; however, mice with a homozygous 5-bp deletion in U2 ortholog Rnu2-8 present with ataxia and neurodegeneration32. Transcriptomic analysis of the mutant cerebellum detected aberrant splicing, particularly increased retention of short introns. Although it remains unclear how this splicing defect might cause neuronal death, it has been hypothesized that premature translation termination codons within the retained introns could trigger the nonsense-mediated decay (NMD) pathway. We and others have shown that the recessive human disorders caused by variants in RNU4ATAC and RNU12 result in minor intron retention in blood cells and fibroblasts2,4,6,33,34. By contrast, we have been unable to detect any significant and reproducible large-scale splicing defect in the blood cells of patients with dominant germline variants in the major spliceosome gene RNU2-2. Although a recent study described systematic disruption of 5′ splice site usage in the whole blood of some patients with de novo RNU4-2 variants10, RNA-seq of fibroblasts in a separate case study could not detect any defect in splicing25. Moreover, transcriptomic analysis of primary hematological tumors and cell lines transfected with vectors expressing the n.28C>T RNU2-2 mutation did not reveal any significant differences in splicing18. Therefore, further studies are required to understand how RNU4-2 and RNU2-2 mutations affect splicing. It might be that, in contrast to recessive splicing disorders, it is challenging to detect widespread splicing defects in these newly discovered dominant disorders because wild-type transcripts are expressed in combination with misspliced transcripts from the same gene that are subjected to NMD. In certain cell types, the effects of NMD might be overcome such that the overall expression levels of mRNAs remain unchanged, owing to rapid mRNA turnover and dosage compensation35. However, certain cell types, such as stem cells, which we have not yet been able to study, might be more sensitive to high NMD dosage than terminally differentiated cells. Neuronal stem cells and mouse models of RNU4-2 and RNU2-2 pathologies may be needed to resolve these mechanistic questions.