Kaschta, D. et al. Evaluating genome sequencing strategies: trio, singleton, and standard testing in rare disease diagnosis. Preprint at medRxiv https://doi.org/10.1101/2024.12.20.24319228 (2024).
Wojcik, M. H. et al. Genome sequencing for diagnosing rare diseases. N. Engl. J. Med. 390, 1985–1997 (2024).
Hodder, A. et al. Benefits for children with suspected cancer from routine whole-genome sequencing. Nat. Med. 30, 1905–1912 (2024).
Kerle, I. A. et al. Translational and clinical comparison of whole genome and transcriptome to panel sequencing in precision oncology. npj Precis. Oncol. 9, 9 (2025).
Ellingford, J. M. et al. Recommendations for clinical interpretation of variants found in non-coding regions of the genome. Genome Med. 14, 73 (2022).
100,000 Genomes Project Pilot Investigatorset al. 100,000 genomes pilot on rare-disease diagnosis in health care — preliminary report. N. Engl. J. Med. 385, 1868–1880 (2021).
Nieboer, M. M., Nguyen, L. & de Ridder, J. Predicting pathogenic non-coding SVs disrupting the 3D genome in 1646 whole cancer genomes using multiple instance learning. Sci. Rep. 11, 14411 (2021).
Pagnamenta, A. T. et al. Structural and non-coding variants increase the diagnostic yield of clinical whole genome sequencing for rare diseases. Genome Med. 15, 94 (2023).
Alkan, C., Coe, B. P. & Eichler, E. E. Genome structural variation discovery and genotyping. Nat. Rev. Genet. 12, 363–376 (2011).
Huddleston, J. et al. Discovery and genotyping of structural variation from long-read haploid genome sequence data. Genome Res. 27, 677–685 (2017).
Audano, P. A. et al. Characterizing the major structural variant alleles of the human genome. Cell 176, 663–675.e19 (2019).
Ebert, P. et al. Haplotype-resolved diverse human genomes and integrated analysis of structural variation. Science 372, eabf7117 (2021).
Collins, R. L. et al. A structural variation reference for medical and population genetics. Nature 581, 444–451 (2020).
Fantes, J. et al. Aniridia-associated cytogenetic rearrangements suggest that a position effect may cause the mutant phenotype. Hum. Mol. Genet. 4, 415–422 (1995).
Lettice, L. A. et al. Disruption of a long-range cis-acting regulator for Shh causes preaxial polydactyly. Proc. Natl Acad. Sci. USA 99, 7548–7553 (2002).
Kleinjan, D. J. & van Heyningen, V. Position effect in human genetic disease. Hum. Mol. Genet. 7, 1611–1618 (1998).
Socha, M. et al. Position effects at the FGF8 locus are associated with femoral hypoplasia. Am. J. Hum. Genet. 108, 1725–1734 (2021). This study demonstrated the differences between gene dosage effects and position effects in individuals with limb malformation and mouse models.
Spielmann, M., Lupiáñez, D. G. & Mundlos, S. Structural variation in the 3D genome. Nat. Rev. Genet. 19, 453–467 (2018).
Kleinjan, D. A. et al. Aniridia-associated translocations, DNase hypersensitivity, sequence comparison and transgenic analysis redefine the functional domain of PAX6. Hum. Mol. Genet. 10, 2049–2059 (2001).
Velagaleti, G. V. et al. Position effects due to chromosome breakpoints that map ~900 Kb upstream and ~1.3 Mb downstream of SOX9 in two patients with campomelic dysplasia. Am. J. Hum. Genet. 76, 652–662 (2005).
Tufarelli, C. et al. Transcription of antisense RNA leading to gene silencing and methylation as a novel cause of human genetic disease. Nat. Genet. 34, 157–165 (2003).
Lupski, J. R. & Stankiewicz, P. T. Genomic Disorders: The Genomic Basis of Disease (Springer Science & Business Media, 2007).
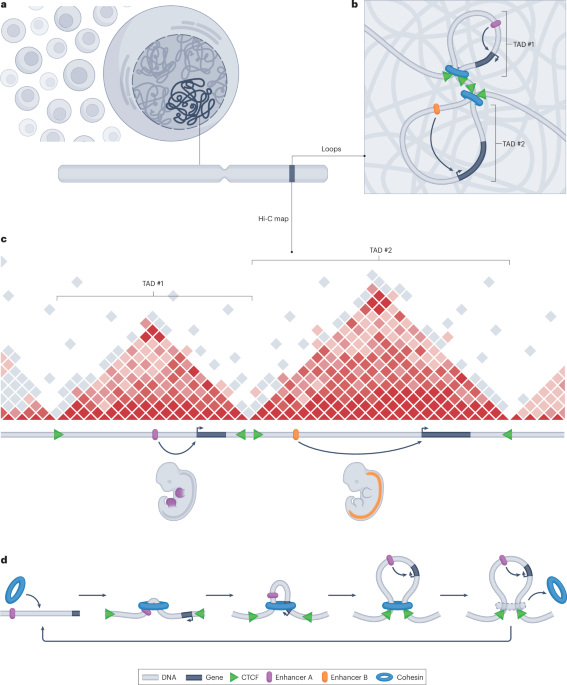
Rao, S. S. P. et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680 (2014).
Buenrostro, J. D., Giresi, P. G., Zaba, L. C., Chang, H. Y. & Greenleaf, W. J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 10, 1213–1218 (2013).
Lupiáñez, D. G. et al. Disruptions of topological chromatin domains cause pathogenic rewiring of gene–enhancer interactions. Cell 161, 1012–1025 (2015). This study introduced the concept of TADs as a useful unit to interpret position effects of SVs caused by altered 3D genome architecture in the context of rare diseases.
Liu, Z. et al. Linking genome structures to functions by simultaneous single-cell Hi-C and RNA-seq. Science 380, 1070–1076 (2023).
Wu, H.-J. et al. Topological isolation of developmental regulators in mammalian genomes. Nat. Commun. 12, 4897 (2021).
Tan, L. et al. Lifelong restructuring of 3D genome architecture in cerebellar granule cells. Science 381, 1112–1119 (2023).
Dehingia, B., Milewska, M., Janowski, M. & Pękowska, A. CTCF shapes chromatin structure and gene expression in health and disease. EMBO Rep. 23, e55146 (2022).
Hnisz, D. et al. Activation of proto-oncogenes by disruption of chromosome neighborhoods. Science 351, 1454–1458 (2016).
Nurk, S. et al. The complete sequence of a human genome. Science 376, 44–53 (2022).
Tchasovnikarova, I. A. et al. Gene silencing. Epigenetic silencing by the HUSH complex mediates position–effect variegation in human cells. Science 348, 1481–1485 (2015).
Davidson, I. F. & Peters, J.-M. Genome folding through loop extrusion by SMC complexes. Nat. Rev. Mol. Cell Biol. 22, 445–464 (2021).
Zhang, D., Lam, J. & Blobel, G. A. Engineering three-dimensional genome folding. Nat. Genet. 53, 602–611 (2021).
Dekker, C., Haering, C. H., Peters, J.-M. & Rowland, B. D. How do molecular motors fold the genome? Science 382, 646–648 (2023).
Zhang, Y. et al. Computational methods for analysing multiscale 3D genome organization. Nat. Rev. Genet. 25, 123–141 (2024).
Jerkovic, I. & Cavalli, G. Understanding 3D genome organization by multidisciplinary methods. Nat. Rev. Mol. Cell Biol. 22, 511–528 (2021).
Raca, G. et al. Points to consider in the detection of germline structural variants using next-generation sequencing: a statement of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 25, 100316 (2023).
Dubois, F., Sidiropoulos, N., Weischenfeldt, J. & Beroukhim, R. Structural variations in cancer and the 3D genome. Nat. Rev. Cancer 22, 533–546 (2022).
Rowley, M. J. & Corces, V. G. Organizational principles of 3D genome architecture. Nat. Rev. Genet. 19, 789–800 (2018).
Schwarzer, W. et al. Two independent modes of chromatin organization revealed by cohesin removal. Nature 551, 51–56 (2017).
Marchal, C., Sima, J. & Gilbert, D. M. Control of DNA replication timing in the 3D genome. Nat. Rev. Mol. Cell Biol. 20, 721–737 (2019).
Arnould, C. & Legube, G. The secret life of chromosome loops upon DNA double-strand break. J. Mol. Biol. 432, 724–736 (2020).
Hill, L. et al. Wapl repression by Pax5 promotes V gene recombination by Igh loop extrusion. Nature 584, 142–147 (2020).
Rajderkar, S. et al. Topologically associating domain boundaries are required for normal genome function. Commun. Biol. 6, 435 (2023).
Oji, A. et al. Structure and dynamics of nuclear A/B compartments and subcompartments. Curr. Opin. Cell Biol. 90, 102406 (2024).
Jabbari, K., Chakraborty, M. & Wiehe, T. DNA sequence-dependent chromatin architecture and nuclear hubs formation. Sci. Rep. 9, 1–11 (2019).
Dixon, J. R. et al. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485, 376–380 (2012). This paper first reported the identification and description of topologically associating domains from Hi-C maps.
Lieberman-Aiden, E. et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326, 289–293 (2009).
Nora, E. P. et al. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature 485, 381–385 (2012).
Dixon, J. R., Gorkin, D. U. & Ren, B. Chromatin domains: the unit of chromosome organization. Mol. Cell 62, 668–680 (2016).
Dekker, J., Marti-Renom, M. A. & Mirny, L. A. Exploring the three-dimensional organization of genomes: interpreting chromatin interaction data. Nat. Rev. Genet. 14, 390–403 (2013).
Zuin, J. et al. Nonlinear control of transcription through enhancer–promoter interactions. Nature 604, 571–577 (2022).
Sanborn, A. L. et al. Chromatin extrusion explains key features of loop and domain formation in wild-type and engineered genomes. Proc. Natl Acad. Sci. USA 112, E6456–E6465 (2015). This study convincingly established a link between the loop extrusion model and TADs based on engineered genomes and polymer-based computational modelling.
Fudenberg, G. et al. Formation of chromosomal domains by loop extrusion. Cell Rep. 15, 2038–2049 (2016).
Oldenkamp, R. & Rowland, B. D. A walk through the SMC cycle: from catching DNAs to shaping the genome. Mol. Cell 82, 1616–1630 (2022).
Valton, A.-L. et al. A cohesin traffic pattern genetically linked to gene regulation. Nat. Struct. Mol. Biol. 29, 1239–1251 (2022).
Arnould, C. et al. Loop extrusion as a mechanism for formation of DNA damage repair foci. Nature 590, 660–665 (2021).
Dequeker, B. J. H. et al. MCM complexes are barriers that restrict cohesin-mediated loop extrusion. Nature 606, 197–203 (2022).
Nanni, L., Ceri, S. & Logie, C. Spatial patterns of CTCF sites define the anatomy of TADs and their boundaries. Genome Biol. 21, 1–25 (2020).
Pradhan, B. et al. SMC complexes can traverse physical roadblocks bigger than their ring size. Cell Rep. 41, 111491 (2022).
Gabriele, M. et al. Dynamics of CTCF- and cohesin-mediated chromatin looping revealed by live-cell imaging. Science 376, 496–501 (2022).
Beagan, J. A. & Phillips-Cremins, J. E. On the existence and functionality of topologically associating domains. Nat. Genet. 52, 8–16 (2020).
Narendra, V. et al. CTCF establishes discrete functional chromatin domains at the Hox clusters during differentiation. Science 347, 1017–1021 (2015).
Rao, S. S. P. et al. Cohesin loss eliminates all loop domains. Cell 171, 305–320.e24 (2017).
Hsieh, T.-H. S. et al. Enhancer–promoter interactions and transcription are largely maintained upon acute loss of CTCF, cohesin, WAPL or YY1. Nat. Genet. 54, 1919–1932 (2022).
Rinzema, N. J. et al. Building regulatory landscapes reveals that an enhancer can recruit cohesin to create contact domains, engage CTCF sites and activate distant genes. Nat. Struct. Mol. Biol. 29, 563–574 (2022).
Calderon, L. et al. Cohesin-dependence of neuronal gene expression relates to chromatin loop length. eLife 11, e76539 (2022).
Kane, L. et al. Cohesin is required for long-range enhancer action at the Shh locus. Nat. Struct. Mol. Biol. 29, 891–897 (2022).
Dong, P. et al. Cohesin prevents cross-domain gene coactivation. Nat. Genet. 56, 1654–1664 (2024).
Akdemir, K. C. et al. Disruption of chromatin folding domains by somatic genomic rearrangements in human cancer. Nat. Genet. 52, 294–305 (2020).
Katainen, R. et al. CTCF/cohesin-binding sites are frequently mutated in cancer. Nat. Genet. 47, 818–821 (2015).
Liu, T. et al. Enhancer coamplification and hijacking promote oncogene expression in liposarcoma. Cancer Res. 83, 1517–1530 (2023).
Zhao, S. G. et al. Integrated analyses highlight interactions between the three-dimensional genome and DNA, RNA and epigenomic alterations in metastatic prostate cancer. Nat. Genet. 56, 1689–1700 (2024).
Soto, D. C. et al. Genomic structural variation: a complex but important driver of human evolution. Am. J. Biol. Anthropol. 181, 118–144 (2023).
Yoo, D. et al. Complete sequencing of ape genomes. Nature 641, 401–418 (2025).
Grimes, K. et al. Cell-type-specific consequences of mosaic structural variants in hematopoietic stem and progenitor cells. Nat. Genet. 56, 1134–1146 (2024).
Stankiewicz, P. & Lupski, J. R. Structural variation in the human genome and its role in disease. Annu. Rev. Med. 61, 437–455 (2010).
Sano, Y. et al. Likely pathogenic structural variants in genetically unsolved patients with retinitis pigmentosa revealed by long-read sequencing. J. Med. Genet. 59, 1133–1138 (2022).
Pagnamenta, A. T. et al. The impact of inversions across 33,924 families with rare disease from a national genome sequencing project. Am. J. Hum. Genet. 111, 1140–1164 (2024).
Bardoni, B. et al. A dosage sensitive locus at chromosome Xp21 is involved in male to female sex reversal. Nat. Genet. 7, 497–501 (1994).
Dong, Y. et al. Targeted next-generation sequencing identification of mutations in patients with disorders of sex development. BMC Med. Genet. 17, 23 (2016).
Holder-Espinasse, M. et al. Duplication of 10q24 locus: broadening the clinical and radiological spectrum. Eur. J. Hum. Genet. 27, 525–534 (2019).
Si, N. et al. Duplications involving the long range HMX1 enhancer are associated with human isolated bilateral concha-type microtia. J. Transl. Med. 18, 244 (2020).
Liu, Y. et al. Germline intergenic duplications at Xq26.1 underlie Bazex–Dupré–Christol basal cell carcinoma susceptibility syndrome. Br. J. Dermatol. 187, 948–961 (2022).
Yumiceba, V. & Spielmann, M. When too much is too much: noncoding duplications in skin disorders. Br. J. Dermatol. 188, e2–e3 (2023).
Pagnamenta, A. T. et al. Conclusion of diagnostic odysseys due to inversions disrupting GLI3 and FBN1. J. Med. Genet. 60, 505–510 (2023).
Lu, B., Jiang, R., Xie, B., Wu, W. & Zhao, Y. Fusion genes in gynecologic tumors: the occurrence, molecular mechanism and prospect for therapy. Cell Death Dis. 12, 783 (2021).
Gryder, B. E. et al. Miswired enhancer logic drives a cancer of the muscle lineage. iScience 23, 101103 (2020).
Baxter, J. S. et al. Functional annotation of the 2q35 breast cancer risk locus implicates a structural variant in influencing activity of a long-range enhancer element. Am. J. Hum. Genet. 108, 1190–1203 (2021).
Ibn-Salem, J. et al. Deletions of chromosomal regulatory boundaries are associated with congenital disease. Genome Biol. 15, 423 (2014).
Lettice, L. A. et al. Enhancer-adoption as a mechanism of human developmental disease. Hum. Mutat. 32, 1492–1499 (2011).
Gröschel, S. et al. A single oncogenic enhancer rearrangement causes concomitant EVI1 and GATA2 deregulation in leukemia. Cell 157, 369–381 (2014).
Redin, C. et al. The genomic landscape of balanced cytogenetic abnormalities associated with human congenital anomalies. Nat. Genet. 49, 36–45 (2017).
Dimartino, P. et al. Structural variants at the LMNB1 locus: deciphering pathomechanisms in autosomal dominant adult-onset demyelinating leukodystrophy. Ann. Neurol. 96, 855–870 (2024).
Northcott, P. A. et al. Enhancer hijacking activates GFI1 family oncogenes in medulloblastoma. Nature 511, 428–434 (2014).
Baudic, M. et al. TAD boundary deletion causes PITX2-related cardiac electrical and structural defects. Nat. Commun. 15, 3380 (2024). These authors showed that deletion of a TAD boundary — identified to be the minimal critical region across seven families with congenital heart defects — leads to enhancer hijacking and altered differentiation during heart development.
Salnikov, P. et al. Structural variants in the Epb41l4a locus: TAD disruption and Nrep gene misregulation as hypothetical drivers of neurodevelopmental outcomes. Sci. Rep. 14, 5288 (2024).
Hirsch, N. et al. HDAC9 structural variants disrupting TWIST1 transcriptional regulation lead to craniofacial and limb malformations. Genome Res. 32, 1242–1253 (2022).
Weischenfeldt, J. et al. Pan-cancer analysis of somatic copy-number alterations implicates IRS4 and IGF2 in enhancer hijacking. Nat. Genet. 49, 65–74 (2017). This report first described the importance of enhancer hijacking in cancer owing to somatic CNVs.
Wang, X. et al. Genome-wide detection of enhancer-hijacking events from chromatin interaction data in rearranged genomes. Nat. Methods 18, 661–668 (2021). This paper reported a computational tool to identify enhancer hijacking events from patient-derived Hi-C maps and its visual representation in the reconstructed genome.
Flöttmann, R. et al. Noncoding copy-number variations are associated with congenital limb malformation. Genet. Med. 20, 599–607 (2018).
Newman, S., Hermetz, K. E., Weckselblatt, B. & Rudd, M. K. Next-generation sequencing of duplication CNVs reveals that most are tandem and some create fusion genes at breakpoints. Am. J. Hum. Genet. 96, 208–220 (2015).
Franke, M. et al. Formation of new chromatin domains determines pathogenicity of genomic duplications. Nature 538, 265–269 (2016).
Franke, M. et al. Duplications disrupt chromatin architecture and rewire GPR101–enhancer communication in X-linked acrogigantism. Am. J. Hum. Genet. 109, 553–570 (2022).
Fishilevich, S. et al. GeneHancer: genome-wide integration of enhancers and target genes in GeneCards. Database 2017, bax028 (2017).
de Bruijn, S. E. et al. Structural variants create new topological-associated domains and ectopic retinal enhancer–gene contact in dominant retinitis pigmentosa. Am. J. Hum. Genet. 107, 802–814 (2020).
Meinel, J. A. et al. Disruption of the topologically associated domain at Xp21.2 is related to 46,XY gonadal dysgenesis. J. Med. Genet. 60, 469–476 (2023).
Veyt, N., Van Buggenhout, G., Devriendt, K., Van Den Bogaert, K. & Brison, N. Expanding the phenotype of copy number variations involving NR0B1 (DAX1). Eur. J. Hum. Genet. 32, 421–425 (2024).
Gardner, J. C. et al. Inter-chromosomal insertions at Xq27.1 associated with retinal dystrophy induce dysregulation of LINC00632 and CDR1as/ciRS-7. Am. J. Hum. Genet. 112, 523–536 (2025).
Melo, U. S. et al. Enhancer hijacking at the ARHGAP36 locus is associated with connective tissue to bone transformation. Nat. Commun. 14, 2034 (2023). This study found that enhancer hijacking resulted in a striking phenotype of ossification of connective tissue with postnatal disease onset.
Ramisch, A. et al. CRUP: a comprehensive framework to predict condition-specific regulatory units. Genome Biol. 20, 227 (2019).
Le Caignec, C. et al. Fryns type mesomelic dysplasia of the upper limbs caused by inverted duplications of the HOXD gene cluster. Eur. J. Hum. Genet. 28, 324–332 (2020).
Lonfat, N. & Duboule, D. Structure, function and evolution of topologically associating domains (TADs) at HOX loci. FEBS Lett. 589, 2869–2876 (2015).
Sun, W. et al. Altered chromatin topologies caused by balanced chromosomal translocation lead to central iris hypoplasia. Nat. Commun. 15, 5048 (2024). This team identified a translocation-led enhancer hijacking by APCDD1 in the developing iris as the molecular diagnosis of central iris hypoplasia in several individuals from a large family.
Creyghton, M. P. et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl Acad. Sci. USA 107, 21931–21936 (2010).
Boix, C. A., James, B. T., Park, Y. P., Meuleman, W. & Kellis, M. Regulatory genomic circuitry of human disease loci by integrative epigenomics. Nature 590, 300–307 (2021).
Damián, A. et al. Long-read genome sequencing identifies cryptic structural variants in congenital aniridia cases. Hum. Genomics 17, 45 (2023).
Yang, L. et al. 3D genome alterations associated with dysregulated HOXA13 expression in high-risk T-lineage acute lymphoblastic leukemia. Nat. Commun. 12, 3708 (2021).
Laugsch, M. et al. Modeling the pathological long-range regulatory effects of human structural variation with patient-specific hiPSCs. Cell Stem Cell 24, 736–752.e12 (2019). This study found that pericentric inversion entails topologically associating domain shuffling and enhancer disconnection, leading to species-specific haploinsufficiency and branchio-oculo-facial syndrome.
Milunsky, J. M. et al. TFAP2A mutations result in branchio-oculo-facial syndrome. Am. J. Hum. Genet. 82, 1171–1177 (2008).
Zhang, J. et al. Neural tube, skeletal and body wall defects in mice lacking transcription factor AP-2. Nature 381, 238–241 (1996).
Schorle, H., Meier, P., Buchert, M., Jaenisch, R. & Mitchell, P. J. Transcription factor AP-2 essential for cranial closure and craniofacial development. Nature 381, 235–238 (1996).
Chandrasekhar, A. et al. Genome sequencing detects a balanced pericentric inversion with breakpoints that impact the DMD and upstream region of POU3F4 genes. Am. J. Med. Genet. A 194, e63462 (2024).
Schöpflin, R. et al. Integration of Hi-C with short and long-read genome sequencing reveals the structure of germline rearranged genomes. Nat. Commun. 13, 6470 (2022).
Ghavi-Helm, Y. et al. Highly rearranged chromosomes reveal uncoupling between genome topology and gene expression. Nat. Genet. 51, 1272–1282 (2019).
Despang, A. et al. Functional dissection of the Sox9-Kcnj2 locus identifies nonessential and instructive roles of TAD architecture. Nat. Genet. 51, 1263–1271 (2019).
Rodríguez-Carballo, E. et al. The HoxD cluster is a dynamic and resilient TAD boundary controlling the segregation of antagonistic regulatory landscapes. Genes Dev. 31, 2264–2281 (2017).
Ge, X. et al. Outward-oriented sites within clustered CTCF boundaries are key for intra-TAD chromatin interactions and gene regulation. Nat. Commun. 14, 8101 (2023).
Mohajeri, K. et al. Transcriptional and functional consequences of alterations to MEF2C and its topological organization in neuronal models. Am. J. Hum. Genet. 109, 2049–2067 (2022).
Kang, J., Kim, Y. W., Park, S., Kang, Y. & Kim, A. Multiple CTCF sites cooperate with each other to maintain a TAD for enhancer–promoter interaction in the β-globin locus. FASEB J. 35, e21768 (2021).
Chang, L.-H. et al. Multi-feature clustering of CTCF binding creates robustness for loop extrusion blocking and topologically associating domain boundaries. Nat. Commun. 14, 5615 (2023).
Kabirova, E. et al. TAD border deletion at the Kit locus causes tissue-specific ectopic activation of a neighboring gene. Nat. Commun. 15, 4521 (2024). This paper describes the tissue specificity of enhancer hijacking and gene misexpression upon boundary deletions based on the interplay among loop extrusion, transcription machinery and compensatory CTCF motifs.
Huang, H. et al. CTCF mediates dosage- and sequence-context-dependent transcriptional insulation by forming local chromatin domains. Nat. Genet. 53, 1064–1074 (2021).
Banigan, E. J. et al. Transcription shapes 3D chromatin organization by interacting with loop extrusion. Proc. Natl Acad. Sci. USA 120, e2210480120 (2023).
Botten, G. A. et al. Structural variation cooperates with permissive chromatin to control enhancer hijacking-mediated oncogenic transcription. Blood 142, 336–351 (2023).
Chakraborty, S. et al. Enhancer–promoter interactions can bypass CTCF-mediated boundaries and contribute to phenotypic robustness. Nat. Genet. 55, 280–290 (2023).
Barshad, G. et al. RNA polymerase II dynamics shape enhancer–promoter interactions. Nat. Genet. 55, 1370–1380 (2023).
Bolt, C. C. et al. Context-dependent enhancer function revealed by targeted inter-TAD relocation. Nat. Commun. 13, 3488 (2022).
Winick-Ng, W. et al. Cell-type specialization is encoded by specific chromatin topologies. Nature 599, 684–691 (2021).
Tian, W. et al. Single-cell DNA methylation and 3D genome architecture in the human brain. Science 382, eadf5357 (2023).
Kraft, K. et al. Serial genomic inversions induce tissue-specific architectural stripes, gene misexpression and congenital malformations. Nat. Cell Biol. 21, 305–310 (2019).
Stadhouders, R. et al. Transcription factors orchestrate dynamic interplay between genome topology and gene regulation during cell reprogramming. Nat. Genet. 50, 238–249 (2018).
Wang, J. et al. Phase separation of OCT4 controls TAD reorganization to promote cell fate transitions. Cell Stem Cell 28, 1868–1883.e11 (2021).
Szabo, Q. et al. Regulation of single-cell genome organization into TADs and chromatin nanodomains. Nat. Genet. 52, 1151–1157 (2020).
Mensah, M. A. et al. Aberrant phase separation and nucleolar dysfunction in rare genetic diseases. Nature 614, 564–571 (2023).
Kragesteen, B. K., Brancati, F., Digilio, M. C., Mundlos, S. & Spielmann, M. H2AFY promoter deletion causes PITX1 endoactivation and Liebenberg syndrome. J. Med. Genet. 56, 246–251 (2019).
Bell, A. C. & Felsenfeld, G. Methylation of a CTCF-dependent boundary controls imprinted expression of the Igf2 gene. Nature 405, 482–485 (2000).
Hark, A. T. et al. CTCF mediates methylation-sensitive enhancer-blocking activity at the H19/Igf2 locus. Nature 405, 486–489 (2000).
Monteagudo-Sánchez, A., Richard, A. J., Scarpa, M., Noordermeer, D. & Greenberg, M. V. C. The impact of the embryonic DNA methylation program on CTCF-mediated genome regulation. Nucleic Acids Res. 52, 10934–10950 (2024).
Hung, T.-C., Kingsley, D. M. & Boettiger, A. N. Boundary stacking interactions enable cross-TAD enhancer–promoter communication during limb development. Nat. Genet. 56, 306–314 (2024). This work presents a revised perspective on the function of TAD boundaries, highlighting their dual role in both facilitating and restricting cis-regulatory interactions.
Yokoshi, M., Segawa, K. & Fukaya, T. Visualizing the role of boundary elements in enhancer–promoter communication. Mol. Cell 78, 224–235.e5 (2020).
Davidson, I. F. et al. CTCF is a DNA-tension-dependent barrier to cohesin-mediated loop extrusion. Nature 616, 822–827 (2023).
Chen, Z. et al. Increased enhancer–promoter interactions during developmental enhancer activation in mammals. Nat. Genet. 56, 675–685 (2024).
Beccari, L. et al. Dbx2 regulation in limbs suggests interTAD sharing of enhancers. Dev. Dyn. 250, 1280–1299 (2021).
Li, X. et al. GAGA-associated factor fosters loop formation in the Drosophila genome. Mol. Cell 83, 1519–1526.e4 (2023).
Hu, Y. et al. Lineage-specific 3D genome organization is assembled at multiple scales by IKAROS. Cell 186, 5269–5289.e22 (2023).
Boyle, S. et al. A central role for canonical PRC1 in shaping the 3D nuclear landscape. Genes Dev. 34, 931–949 (2020).
Pachano, T., Crispatzu, G. & Rada-Iglesias, A. Polycomb proteins as organizers of 3D genome architecture in embryonic stem cells. Brief. Funct. Genomics 18, 358–366 (2019).
Weiner, D. J. et al. Statistical and functional convergence of common and rare genetic influences on autism at chromosome 16p. Nat. Genet. 54, 1630–1639 (2022).
Loviglio, M. N. et al. Chromosomal contacts connect loci associated with autism, BMI and head circumference phenotypes. Mol. Psychiatry 22, 836–849 (2017).
Zhang, X. et al. Local and global chromatin interactions are altered by large genomic deletions associated with human brain development. Nat. Commun. 9, 5356 (2018).
Konrad, E. D. H. et al. CTCF variants in 39 individuals with a variable neurodevelopmental disorder broaden the mutational and clinical spectrum. Genet. Med. 21, 2723–2733 (2019).
Gregor, A. et al. De novo mutations in the genome organizer CTCF cause intellectual disability. Am. J. Hum. Genet. 93, 124–131 (2013).
Zhang, J. et al. CTCF mutation at R567 causes developmental disorders via 3D genome rearrangement and abnormal neurodevelopment. Nat. Commun. 15, 5524 (2024).
Krantz, I. D. et al. Cornelia de Lange syndrome is caused by mutations in NIPBL, the human homolog of Drosophila melanogaster Nipped-B. Nat. Genet. 36, 631–635 (2004).
Chen, W. et al. Machine learning enables pan-cancer identification of mutational hotspots at persistent CTCF binding sites. Nucleic Acids Res. 52, 8086–8099 (2024).
Bell, A. C., West, A. G. & Felsenfeld, G. The protein CTCF is required for the enhancer blocking activity of vertebrate insulators. Cell 98, 387–396 (1999).
Allou, L. et al. Non-coding deletions identify Maenli lncRNA as a limb-specific En1 regulator. Nature 592, 93–98 (2021).
Quinodoz, M. et al. De novo and inherited dominant variants in U4 and U6 snRNAs cause retinitis pigmentosa. Preprint at medRxiv https://doi.org/10.1101/2025.01.06.24317169 (2025).
Chen, Y. et al. De novo variants in the RNU4-2 snRNA cause a frequent neurodevelopmental syndrome. Nature 632, 832–840 (2024).
FitzPatrick, D. R. et al. Transcriptome analysis of human autosomal trisomy. Hum. Mol. Genet. 11, 3249–3256 (2002).
Meharena, H. S. et al. Down-syndrome-induced senescence disrupts the nuclear architecture of neural progenitors. Cell Stem Cell 29, 116–130.e7 (2022).
He, S. et al. The role of cardiomyocyte senescence in cardiovascular diseases: a molecular biology update. Eur. J. Pharmacol. 983, 176961 (2024).
Mahmood, S. R. et al. β-actin dependent chromatin remodeling mediates compartment level changes in 3D genome architecture. Nat. Commun. 12, 5240 (2021).
Battirossi, E. et al. Assessment of the cytoskeletal impact of beta-actin mutations leading to non-muscle actinopathies by means of dual laser optical tweezers (DLOT). Biophys. J. 122, 195a (2023).
Riggs, E. R. et al. Technical standards for the interpretation and reporting of constitutional copy-number variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet. Med. 22, 245–257 (2020).
McArthur, E. & Capra, J. A. Topologically associating domain boundaries that are stable across diverse cell types are evolutionarily constrained and enriched for heritability. Am. J. Hum. Genet. 108, 269–283 (2021).
Zhang, S., Übelmesser, N., Barbieri, M. & Papantonis, A. Enhancer–promoter contact formation requires RNAPII and antagonizes loop extrusion. Nat. Genet. 55, 832–840 (2023).
ENCODE Project Consortium et al. Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature 583, 699–710 (2020).
Nieboer, M. M. & de Ridder, J. svMIL: predicting the pathogenic effect of TAD boundary-disrupting somatic structural variants through multiple instance learning. Bioinformatics 36, i692–i699 (2020).
Hertzberg, J., Mundlos, S., Vingron, M. & Gallone, G. TADA — a machine learning tool for functional annotation-based prioritisation of pathogenic CNVs. Genome Biol. 23, 67 (2022).
Sharo, A. G., Hu, Z., Sunyaev, S. R. & Brenner, S. E. StrVCTVRE: a supervised learning method to predict the pathogenicity of human genome structural variants. Am. J. Hum. Genet. 109, 195–209 (2022).
Xu, Z., Li, Q., Marchionni, L. & Wang, K. PhenoSV: interpretable phenotype-aware model for the prioritization of genes affected by structural variants. Nat. Commun. 14, 7805 (2023).
Geoffroy, V. et al. AnnotSV and knotAnnotSV: a web server for human structural variations annotations, ranking and analysis. Nucleic Acids Res. 49, W21–W28 (2021).
Fino, J., Marques, B., Dong, Z. & David, D. SVInterpreter: a comprehensive topologically associated domain-based clinical outcome prediction tool for balanced and unbalanced structural variants. Front. Genet. 12, 757170 (2021).
Requena, F. et al. CNVxplorer: a web tool to assist clinical interpretation of CNVs in rare disease patients. Nucleic Acids Res. 49, W93–W103 (2021).
Zhang, L. et al. X-CNV: genome-wide prediction of the pathogenicity of copy number variations. Genome Med. 13, 132 (2021).
Kleinert, P. & Kircher, M. A framework to score the effects of structural variants in health and disease. Genome Res. 32, 766–777 (2022).
Spector, J. D. & Wiita, A. P. ClinTAD: a tool for copy number variant interpretation in the context of topologically associated domains. J. Hum. Genet. 64, 437–443 (2019).
Poszewiecka, B. et al. TADeus2: a web server facilitating the clinical diagnosis by pathogenicity assessment of structural variations disarranging 3D chromatin structure. Nucleic Acids Res. 50, W744–W752 (2022).
Sánchez-Gaya, V. & Rada-Iglesias, A. POSTRE: a tool to predict the pathological effects of human structural variants. Nucleic Acids Res. 51, e54 (2023). This report presented a computational tool for prioritizing a list of SVs, which considers both linear and 3D genome effects in a tissue-specific manner based on clinical phenotype.
Zhou, J. Sequence-based modeling of three-dimensional genome architecture from kilobase to chromosome scale. Nat. Genet. 54, 725–734 (2022).
Melo, U. S. et al. Hi-C identifies complex genomic rearrangements and TAD-shuffling in developmental diseases. Am. J. Hum. Genet. 106, 872–884 (2020). This study introduced Hi-C as a diagnostic tool for analysing clinical samples from various tissues, including blood, amnion and fibroblasts.
Beagrie, R. A. et al. Multiplex-GAM: genome-wide identification of chromatin contacts yields insights overlooked by Hi-C. Nat. Methods 20, 1037–1047 (2023).
Zhong, J.-Y. et al. High-throughput pore-C reveals the single-allele topology and cell type-specificity of 3D genome folding. Nat. Commun. 14, 1250 (2023).
Zhou, T. et al. GAGE-seq concurrently profiles multiscale 3D genome organization and gene expression in single cells. Nat. Genet. 56, 1701–1711 (2024).
Chang, L. et al. Droplet Hi-C enables scalable, single-cell profiling of chromatin architecture in heterogeneous tissues. Nat. Biotechnol. https://doi.org/10.1038/s41587-024-02447-1 (2024).
Yang, R. et al. Epiphany: predicting Hi-C contact maps from 1D epigenomic signals. Genome Biol. 24, 134 (2023).
Abbas, A. et al. ChIPr: accurate prediction of cohesin-mediated 3D genome organization from 2D chromatin features. Genome Biol. 25, 15 (2024).
Schuette, G., Lao, Z. & Zhang, B. ChromoGen: diffusion model predicts single-cell chromatin conformations. Sci. Adv. 11, eadr8265 (2025).
Wang, S. et al. The 3D genome and its impacts on human health and disease. Life Med. 2, lnad012 (2023).
Hoy, S. M. Exagamglogene autotemcel: first approval. Mol. Diagn. Ther. 28, 133–139 (2024).
Frangoul, H. et al. CRISPR–Cas9 gene editing for sickle cell disease and β-thalassemia. N. Engl. J. Med. 384, 252–260 (2021).
Ye, L. et al. Genome editing using CRISPR–Cas9 to create the HPFH genotype in HSPCs: an approach for treating sickle cell disease and β-thalassemia. Proc. Natl Acad. Sci. USA 113, 10661–10665 (2016).
Himadewi, P. et al. 3’HS1 CTCF binding site in human β-globin locus regulates fetal hemoglobin expression. eLife 10, e70557 (2021).
Pinglay, S. et al. Multiplex generation and single-cell analysis of structural variants in mammalian genomes. Science 387, eado5978 (2025).
Koeppel, J. et al. Randomizing the human genome by engineering recombination between repeat elements. Science 387, eado3979 (2025). Together with Pinglay et al. (2025), this paper used high-throughput technology for multiplexed testing of SVs in mammalian genomes.
Liao, W.-W. et al. A draft human pangenome reference. Nature 617, 312–324 (2023).
Gao, Y. et al. A pangenome reference of 36 Chinese populations. Nature 619, 112–121 (2023).
All of Us Research Program Genomics Investigators. Genomic data in the All of Us research program. Nature 627, 340–346 (2024).
Wang, Y. et al. The 3D Genome Browser: a web-based browser for visualizing 3D genome organization and long-range chromatin interactions. Genome Biol. 19, 151 (2018).
Krietenstein, N. et al. Ultrastructural details of mammalian chromosome architecture. Mol. Cell 78, 554–565.e7 (2020).
Kosicki, M. et al. VISTA Enhancer browser: an updated database of tissue-specific developmental enhancers. Nucleic Acids Res. 53, D324–D330 (2024). This study reports the most extensive collection of experimentally validated tissue-specific enhancers in transgenic mice.
Mills, C., Marconett, C. N., Lewinger, J. P. & Mi, H. PEACOCK: a machine learning approach to assess the validity of cell type-specific enhancer–gene regulatory relationships. npj Syst. Biol. Appl. 9, 9 (2023).
Li, M. et al. Comprehensive 3D epigenomic maps define limbal stem/progenitor cell function and identity. Nat. Commun. 13, 1293 (2022).
Regev, A. et al. The Human Cell Atlas. eLife 6, e27041 (2017).
Cao, J. et al. The single-cell transcriptional landscape of mammalian organogenesis. Nature 566, 496–502 (2019).
Cao, J. et al. A human cell atlas of fetal gene expression. Science 370, eaba7721 (2020).
Zepeda-Mendoza, C. J. & Morton, C. C. The iceberg under water: unexplored complexity of chromoanagenesis in congenital disorders. Am. J. Hum. Genet. 104, 565–577 (2019).
Baca, S. C. et al. Punctuated evolution of prostate cancer genomes. Cell 153, 666–677 (2013).
Stephens, P. J. et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 144, 27–40 (2011).
Liu, P. et al. Chromosome catastrophes involve replication mechanisms generating complex genomic rearrangements. Cell 146, 889–903 (2011).
Li, Y. et al. Patterns of somatic structural variation in human cancer genomes. Nature 578, 112–121 (2020).
Dileep, V. et al. Neuronal DNA double-strand breaks lead to genome structural variations and 3D genome disruption in neurodegeneration. Cell 186, 4404–4421.e20 (2023).
Simovic-Lorenz, M. & Ernst, A. Chromothripsis in cancer. Nat. Rev. Cancer 25, 79–92 (2025).
Xie, T. et al. Pervasive structural heterogeneity rewires glioblastoma chromosomes to sustain patient-specific transcriptional programs. Nat. Commun. 15, 3905 (2024).
Helmsauer, K. et al. Enhancer hijacking determines extrachromosomal circular MYCN amplicon architecture in neuroblastoma. Nat. Commun. 11, 5823 (2020).
Sarni, D. et al. 3D genome organization contributes to genome instability at fragile sites. Nat. Commun. 11, 3613 (2020).
Sidiropoulos, N. et al. Somatic structural variant formation is guided by and influences genome architecture. Genome Res. 32, 643–655 (2022).
Kloosterman, W. P. et al. Chromothripsis as a mechanism driving complex de novo structural rearrangements in the germline. Hum. Mol. Genet. 20, 1916–1924 (2011).
Anderson, N. D. et al. Rearrangement bursts generate canonical gene fusions in bone and soft tissue tumors. Science 361, eaam8419 (2018).
Tamura, T. et al. Breakpoint analysis for cytogenetically balanced translocation revealed unexpected complex structural abnormalities and suggested the position effect for MEF2C. Am. J. Med. Genet. Part A 191, 1632–1638 (2023).
Middelkamp, S. et al. Molecular dissection of germline chromothripsis in a developmental context using patient-derived iPS cells. Genome Med. 9, 9 (2017).
Ordulu, Z. et al. Structural chromosomal rearrangements require nucleotide-level resolution: lessons from next-generation sequencing in prenatal diagnosis. Am. J. Hum. Genet. 99, 1015–1033 (2016).
Melo, U. S. et al. Complete lung agenesis caused by complex genomic rearrangements with neo-TAD formation at the SHH locus. Hum. Genet. 140, 1459–1469 (2021).
Song, F., Xu, J., Dixon, J. & Yue, F. Analysis of Hi-C data for discovery of structural variations in cancer. Methods Mol. Biol. 2301, 143–161 (2022).
Dixon, J. R. et al. Integrative detection and analysis of structural variation in cancer genomes. Nat. Genet. 50, 1388–1398 (2018).